一种磁性表面分子印迹聚合物及其制备方法和应用与流程
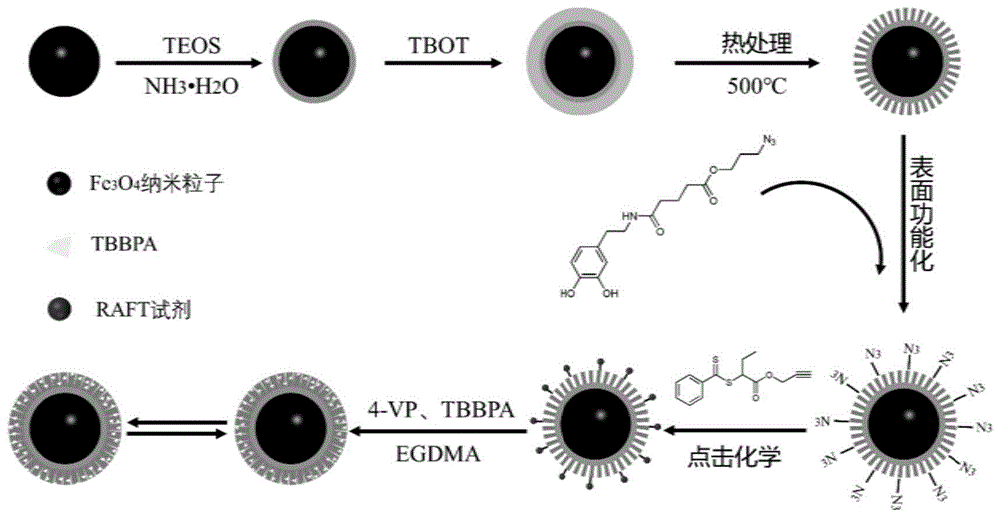
本发明属于分子印迹聚合物技术领域,特别涉及一种磁性表面分子印迹聚合物及其制备方法和应用。
背景技术:
分子印迹聚合物(mips)是一种对目标化合物具有特殊亲和力和特异性识别功能的受体。由于其特异识别功能,mips在化学/生物传感器领域有着广泛的应用。本体聚合是制备分子印迹聚合物的传统方法,该方法所制备的分子印迹聚合物往往要经过研磨、筛分等步骤,制备过程繁琐,而且大量的识别位点被包埋,识别效率低,同时使用过程中存在模板分子泄露的风险,影响检测结果。
表面印迹指的是在固相基质表面制备的印迹聚合物,相比于传统的本体聚合制备的印迹聚合物,表面印迹聚合物具有识别位点容易获得、识别效率高、容易再生、无模板分子泄露等诸多优点,磁性纳米颗粒由于高比表面积、表面易于功能化、易于分离回收等优点成为表面印迹聚合的理想载体,介孔层的引入使得磁性内核易被功能化,并且提供了更加丰富的识别位点。而现有表面印迹技术中印迹载体的比表面积有限,聚合物制备方法为传统自由基聚合,聚合过程副反应多,可控性差,而且难于后修饰。
技术实现要素:
针对现有技术中存在的不足及缺陷,本发明的目的在于提出一种磁性表面分子印迹聚合物及其制备方法和应用,以解决现有技术中表面分子印迹聚合物识别位点有限,分离回收难度大的技术问题。
本发明提出以下的技术方案:
本发明提供了一种磁性表面分子印迹聚合物,以磁性介孔二氧化钛为载体,首先通过叠氮功能化的多巴胺对磁性介孔载体进行表面修饰,然后经点击化学将raft试剂修饰到经过叠氮功能化的多巴胺处理过的磁性介孔载体表面,最后经raft聚合反应,得到所述的磁性表面分子印迹聚合物。
本发明还提供了一种磁性表面分子印迹聚合物的制备方法,包括以下步骤:
步骤1、制备fe3o4磁性纳米颗粒;
步骤2、制备包覆二氧化硅的磁性纳米微球;
步骤3、在磁性纳米微球的表面包覆二氧化钛,得到包覆二氧化钛的磁性纳米微球,然后经磁分离,洗涤,干燥及热处理后,得到磁性介孔二氧化钛纳米微球;
步骤4、对磁性介孔二氧化钛纳米微球进行表面叠氮化处理,得到叠氮功能化的磁性介孔载体;
步骤5、对叠氮功能化的磁性介孔载体进行raft功能化处理,得到raft试剂修饰的磁性介孔二氧化钛载体;
步骤6、将模板分子和功能单体混合均匀,然后加入raft试剂修饰的磁性介孔二氧化钛载体、交联剂及引发剂,使其发生聚合反应,得到磁性介孔分子印迹聚合物;
步骤7、去除磁性介孔分子印迹聚合物中的模板分子,得到所述的磁性表面分子印迹聚合物fe3o4@mtio2@mip。
进一步的,步骤6中,模板分子采用四溴双酚a,功能单体为4-乙烯基吡啶,交联剂为乙二醇二甲基丙烯酸酯,引发剂为偶氮二异丁腈。
进一步的,步骤6中,4-乙烯基吡啶与乙二醇二甲基丙烯酸酯的摩尔比为1:(1-5)。
进一步的,步骤2中,包覆二氧化硅的磁性纳米微球采用溶胶-凝胶法制备得到;具体包括以下步骤:将fe3o4磁性纳米颗粒超声分散在无水乙醇中,加入氨水,超声分散0.5-1h,加入正硅酸乙酯的乙醇溶液;室温条件下,进行反应后,磁分离,洗涤,真空干燥,得到包覆二氧化硅的磁性纳米微球。
进一步的,步骤3中,磁性介孔二氧化钛纳米微球的制备方法为:将包覆二氧化硅的磁性纳米微球分散于无水乙醇中,加入氨水,超声混合;滴加钛酸四丁酯,搅拌反应,得到反应产物;并对反应产物进行磁分离,洗涤,真空干燥,热处理得到磁性介孔二氧化钛纳米微球。
进一步的,步骤4中,叠氮功能化的磁性介孔载体的制备方法为:将磁性介孔二氧化钛纳米微球超声分散至二氯甲烷中,加入叠氮化功能的多巴胺,室温搅拌反应后,磁分离,洗涤,真空干燥,得到叠氮功能化的磁性介孔载体。
进一步的,步骤5中,raft试剂功能化磁性介孔载体的制备方法为:将叠氮功能化的磁性介孔载体超声分散于二甲基亚砜溶液中,加入炔基化raft试剂,分散后,依次加入五水硫酸铜溶液和抗坏血酸钠溶液,进行反应,得到反应产物;对反应产物进行磁分离,洗涤,真空干燥,得到raft试剂修饰的磁性介孔二氧化钛载体。
进一步的,步骤6中,基于磁性表面分子印迹聚合物的制备方法为:将4-乙烯基吡啶与tbbpa溶于无水甲苯中,在氮气的保护下,搅拌,得到混合溶液;将raft试剂修饰的磁性介孔二氧化钛载体,分散于混合溶液中,加入乙二醇二甲基丙烯酸酯和偶氮二异丁腈,在氮气保护下,进行聚合反应,得到反应产物;聚合反应温度为60-70℃,反应时间为16-32h;洗涤,真空干燥,得到磁性介孔分子印迹聚合物。
本发明还提供了一种磁性表面分子印迹聚合物的应用,利用所述的磁性表面分子印迹聚合物在环境体系中对四溴双酚a的识别与检测。
与现有技术相比,本发明具有的有益效果:
本发明提供了一种磁性表面分子印迹聚合物,通过在包覆介孔二氧化钛层的磁性纳米微球表面通过非共价键引入叠氮基,通过点击化学连接raft试剂,通过表面引发的可逆加成-断裂链转移活性自由基聚合反应制备分子印迹聚合物层;制备的磁性表面分子印迹聚合物表现出规则的球形结构且分散性良好并具有明显的多层核壳结构。
本发明还提供了一种磁性表面分子印迹聚合物的制备方法,通过采用fe3o4磁性纳米颗粒作为磁核,方便印迹材料在使用过程中从介质中分离出来;通过在磁核的表面包覆二氧化硅层能有效防止磁核被氧化或者腐蚀;介孔二氧化钛层的引入使得在磁性纳米粒子表面引入叠氮基,介孔结构使识别位点更加丰富,通过在包覆介孔二氧化钛层的磁性纳米微球表面通过非共价键引入叠氮基,通过点击化学连接raft试剂,通过表面引发的可逆加成-断裂链转移活性自由基聚合反应制备分子印迹聚合物层。
进一步的,功能单体与交联剂的比例直接影响分子印迹聚合物的形态和识别性能,本发明中功能单体为4-乙烯基吡啶,交联剂为乙二醇二甲基丙烯酸酯;制备得到的磁性表面分子印迹聚合物表现出最佳的特异性识别效果,功能单体与交联剂之间的很好的协同作用产生了适当交联度并具有一定刚性的印迹识别腔,同时模板分子也容易被去除,进而使磁性表面分子印迹聚合物达到对于模板分子的特异性识别。
本发明还提供了一种磁性表面分子印迹聚合物的应用,由于本发明所述的磁性表面分子印迹聚合物中的印迹层分布于载体表面,使得印迹位点容易接近,达到迅速识别模板分子的效果,提高传质速率。环境体系中对四溴双酚a的识别与检测应用中,实现了有效选择性能和可回收的结果,确保了在复杂环境中对四溴双酚a进行分离。
附图说明
图1是本发明所述的磁性表面分子印迹聚合物的制备过程示意图;
图2是本发明磁性微球表面分子印迹聚合物fe3o4@mtio2@mip的透射电子显微镜照片;
图3是本发明实施例1中制得的fe3o4磁性纳米颗粒(a)、包覆二氧化硅后的磁性纳米微球fe3o4@sio2(b)、磁性介孔二氧化钛纳米微球fe3o4@mtio2(c)、叠氮功能化的磁性纳米介孔载体fe3o4@mtio2-n3(d)、raft试剂修饰后的磁性介孔二氧化钛载体fe3o4@mtio2-cpdb(e)以及磁性表面分子印迹聚合物fe3o4@mtio2@mip(f)的红外光谱图;
图4是本发明实施例1中制得的fe3o4磁性纳米颗粒(a)、包覆二氧化硅后的磁性纳米微球fe3o4@sio2(b)、磁性介孔二氧化钛纳米微球fe3o4@mtio2(c)以及磁性表面分子印迹聚合物fe3o4@mtio2@mip(d)的x射线衍射谱图;
图5是本发明实施例1中制得的fe3o4磁性纳米颗粒(a)、包覆二氧化硅后的磁性纳米微球fe3o4@sio2(b)、磁性介孔二氧化钛纳米微球fe3o4@mtio2(c)以及磁性表面分子印迹聚合物fe3o4@mtio2@mip(d)的磁滞回线。
具体实施方式
下面及实施例对本发明作进一步详细说明,所述是对本发明的解释而不是限定。
本发明提供了一种磁性表面分子印迹聚合物,以磁性介孔二氧化钛为载体,首先通过叠氮功能化的多巴胺对磁性介孔二氧化钛载体进行表面修饰,然后经点击化学将raft试剂修饰在经过叠氮功能化的多巴胺处理过的磁性介孔二氧化钛载体表面,最后经raft聚合反应,得到所述的磁性表面分子印迹聚合物。
本发明还提供了一种磁性表面分子印迹聚合物的制备方法,包括以下步骤:
步骤1、通过溶剂热法制备得到fe3o4磁性纳米颗粒;
步骤2、通过溶胶-凝胶法在fe3o4磁性纳米颗粒表面包覆二氧化硅,得到包覆二氧化硅的磁性纳米微球fe3o4@sio2;
步骤3、将包覆二氧化硅的磁性纳米微球fe3o4@sio2分散到无水乙醇中,加入氨水和钛酸四丁酯,进行水解反应,得到包覆二氧化钛的磁性纳米微球fe3o4@tio2;然后经磁分离,洗涤,干燥及热处理,得到磁性介孔二氧化钛纳米微球fe3o4@mtio2;
步骤4、将磁性介孔二氧化钛纳米微球fe3o4@mtio2超声分散至无水二氯甲烷中,加入叠氮功能化的多巴胺,室温条件下搅拌,得到叠氮功能化的磁性介孔载体fe3o4@mtio2-n3;
步骤5、将叠氮功能化的磁性介孔载体fe3o4@mtio2-n3分散至n,n-二甲基亚砜中,加入炔基化raft试剂,分散后,依次加入五水硫酸铜溶液和抗坏血酸钠溶液,在氮气保护下,搅拌反应,得到raft试剂修饰的磁性介孔二氧化钛载体fe3o4@mtio2-cpdb;
步骤6、将模板分子和功能单体加入到无水甲苯或乙腈中,混合均匀后,加入raft试剂修饰的磁性介孔二氧化钛载体fe3o4@mtio2-cpdb、交联剂及引发剂,通入氮气除氧,在氮气气氛下,进行聚合反应,聚合反应温度为60-70℃,反应时间为16-32h,得到磁性介孔分子印迹聚合物;
步骤7、去除磁性介孔分子印迹聚合物中的模板分子,得到所述的磁性表面分子印迹聚合物fe3o4@mtio2@mip。
本发明所述的磁性表面分子印迹聚合物的制备方法,通过采用溶剂热法制备的fe3o4磁性纳米颗粒作为磁核,使用过程中能够方便将磁性表面分子印迹聚合物从介质中分离出来;通过在fe3o4磁性纳米颗粒的表面包覆二氧化硅层,有效防止磁核被氧化或者腐蚀;通过在包覆二氧化硅的磁性纳米微球fe3o4@sio2的表面引入介孔二氧化钛层,确保了在磁性介孔二氧化钛纳米微球fe3o4@mtio2的表面通过非共价键引入叠氮基,进而确保了叠氮功能化的磁性介孔载体fe3o4@mtio2-n3的表面具有介孔结构,使得其识别位点更加丰富。
本发明步骤5中通过点击化学将raft试剂装饰在叠氮功能化的磁性介孔载体fe3o4@mtio2-n3,然后通过表面引发的可逆加成-断裂链转移活性自由基聚合反应制备分子印迹聚合物层。
本发明步骤6中的聚合反应过程中,功能单体与交联剂的比例直接影响分子印迹聚合物的形态和识别性能,本发明中功能单体为4-乙烯基吡啶,交联剂为乙二醇二甲基丙烯酸酯;按摩尔比计,4-乙烯基吡啶和乙二醇二甲基丙烯酸酯的比例为1:(1-5);优选的,按摩尔比计,4-乙烯基吡啶和乙二醇二甲基丙烯酸酯比例为1:3,此比例下功能单体与交联剂之间的很好的协同作用产生了适当交联度并具有一定刚性的印迹识别腔,同时模板分子也容易被去除,进而使磁性表面分子印迹聚合物达到对于模板分子的快速传质和特异性识别。
实施例1
本实施例对磁性表面分子印迹聚合物的制备方法进行详细说明,如附图1所示,附图1为本发明所述的磁性表面分子印迹聚合物的制备流程示意图;本实施例中具体步骤如下:
步骤1、采用溶剂热法制备fe3o4磁性纳米颗粒;
具体的,首先,称取2.6g无水三氯化铁、1.0g柠檬酸钠及4.0g无水乙酸钠;将称取的无水三氯化铁、柠檬酸钠及无水乙酸钠溶解于80ml乙二醇中,超声15-30min,使其充分溶解,得到均匀溶液;
然后,将上述均匀溶液倒入不锈钢高压釜中,进行反应;其中,反应温度为180-200℃,反应时间为10-24h;反应完成后,冷却至室温,得到黑色产物;
最后,依次采用乙醇和去离子水对黑色产物进行洗涤至上清液澄清,真空干燥,得到fe3o4磁性纳米颗粒;真空干燥温度为40-60℃,真空干燥时间为4-6h。
步骤2、采用溶胶-凝胶法在fe3o4磁性纳米颗粒表面包覆二氧化硅层,得到包覆二氧化硅的磁性纳米微球fe3o4@sio2;
具体的,首先,将0.2-0.4gfe3o4磁性纳米颗粒超声分散到乙醇溶液中,其中,乙醇溶液包括30ml的乙醇和10ml水;加入1.2ml的氨水,继续超声分散0.5-1h,得到均匀分散体系;
然后,在均匀分散体系中加入正硅酸乙酯,室温条件下,反应4-6h后,磁分离,洗涤,真空干燥,得到包覆二氧化硅的磁性纳米微球fe3o4@sio2;洗涤过程依次采用乙醇和去离子水洗涤三次;真空干燥的温度为40-60℃,真空干燥时间为4-6h。
步骤3、制备磁性介孔二氧化钛纳米微球fe3o4@mtio2;
具体的,称取0.2g包覆二氧化硅的磁性纳米微球fe3o4@sio2分散于100ml无水乙醇中,加入0.2ml氨水,超声混合15min,滴加0.75ml钛酸四丁酯,在温度为45-60℃条件下,机械搅拌反应12-24h,得到反应产物;并对反应产物进行磁分离,洗涤,真空干燥,在温度500℃条件下,进行热处理,热处理时间为2-3h,得到磁性介孔二氧化钛纳米微球fe3o4@mtio2;其中,洗涤过程依次采用乙醇和去离子水洗涤三次;真空干燥的温度为40-60℃,真空干燥时间为4-6h。
步骤4、制备叠氮功能化的磁性介孔载体fe3o4@mtio2-n3;
具体的,首先称取0.2g的磁性介孔二氧化钛纳米微球fe3o4@mtio2超声分散至100ml二氯甲烷中,然后加入0.1g叠氮化功能的多巴胺,室温条件下,搅拌反应24-48h后;磁分离,洗涤,真空干燥,得到叠氮功能化的磁性介孔载体fe3o4@mtio2-n3;其中,采用二氯甲烷进行洗涤,洗涤次数为3-5次;真空干燥的温度为40-60℃,真空干燥时间为4-6h。
其中,叠氮化功能的多巴胺的制备方法,包括以下步骤:
s1、在0℃条件下,将3.76g的叠氮化钠加入到2.6ml的3-溴-1-丙醇溶液中,搅拌,加热至60-80℃,保温24-30h,冷却至室温后,采用二氯甲烷萃取,得到萃取液;
s2、向萃取液中加入无水硫酸镁干燥,并通过旋转蒸发去除溶剂,得到化合物一;
s3、将3g化合物一、3.38g戊二酸酐和0.272g4-二甲氨基吡啶溶解在50ml无水二氯甲烷溶液中,得到混合物;将混合物在室温条件下,剧烈搅拌12小时,得到混合物溶液;
s4、采用30ml0.5m的盐酸和15ml饱和食盐水依次洗涤混合物溶液后,加入无水硫酸镁进行干燥,减压除去溶剂,并采用柱色谱进行纯化处理,得到无色油状物的化合物二;其中,柱色谱采用sio2/二氯甲烷/甲醇20:1;
s5、将3.34g的化合物二和2.64g的n-羟基琥珀酰亚胺溶解在50ml无水二氯甲烷中,得到混合溶液;将混合溶液冷却至-5-0℃;将二环己基碳二亚胺的二氯甲烷溶液滴加到上述混合溶液中,升温至室温条件下,搅拌12h,过滤,蒸发溶剂,并采用柱色谱二进行纯化,得到无色油状物的化合物三;其中,二环己基碳二亚胺的二氯甲烷溶液采用将2.68g的二环己基碳二亚胺与10ml的二氯甲烷配制得到;柱色谱二采用sio2/二氯甲烷/甲醇40:1;
s6、将2.35g的多巴胺盐酸盐溶解在50ml的甲醇中,加入1.55g三乙胺,加入化合物三的甲醇溶液,剧烈搅拌反应48-56h,减压蒸发溶剂后,得到残留物;其中,化合物三的甲醇溶液采用将4.8g的化合物三溶解在20ml的甲醇中配制得到;将残留物溶解在二氯甲烷中,采用0.5m的盐酸洗涤有机相后,加入无水硫酸镁干燥,过滤,蒸发溶剂,采用柱色谱三对粗产物进行纯化,得到浅棕色固体,即为叠氮功能化的多巴胺;其中,柱色谱三采用sio2/二氯甲烷/甲醇20:1。
步骤5、制备raft试剂修饰的磁性介孔二氧化钛载体fe3o4@mtio2-cpdb;
具体的,将80mg叠氮功能化的磁性介孔载体fe3o4@mtio2-n3超声分散于25ml二甲基亚砜中,加入40mg炔基化raft试剂,分散15-30min后,依次加入五水硫酸铜溶液和抗坏血酸钠溶液,进行反应,得到反应产物;其中,五水硫酸铜溶液的浓度为0.53mmol/l,抗坏血酸钠溶液的浓度为2.66mmol/l;反应温度为50-60℃,反应时间为48-56h;对反应产物进行磁分离,洗涤,真空干燥,得到raft试剂修饰的磁性介孔二氧化钛载体fe3o4@mtio2-cpdb;洗涤时依次采用去离子水和乙醇洗涤,真空干燥的温度为40-60℃,真空干燥时间为4-6h;
其中,炔基化raft试剂的制备方法,包括以下步骤:
首先,将8.0ml的苯基溴化镁溶解在25.0ml的四氢呋喃中,在45℃的油浴条件下搅拌15-30min;加入8.0ml的二硫化碳,保温2h后冷却至室温,得到混合溶液;
然后,在氮气保护下,在混合溶液中加入7.5ml的2-溴丁酸,搅拌反应48-60h,然后加入约10ml的1m的盐酸终止反应,得到反应产物;
其次,采用30.0ml的乙醚对反应产物进行萃取,得到萃取物,萃取物即为红色油状产物2-二硫代苯甲酸丁酸酯(cpdb);
最后,将0.650ml的萃取物、0.783g的1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、0.500g的4-二甲氨基吡啶和10.0ml二氯甲烷加入到烧瓶中,在氮气保护下,搅拌;之后加入0.5ml炔丙醇,在室温下搅拌反应48-60h后,洗涤,萃取,干燥,过滤,减压浓缩后,采用柱色谱四纯化,得到油状残余物,油状残余物即为炔基化的raft试剂;其中,洗涤过程中依次采用100ml的10%碳酸氢钠溶液、100ml水及100ml饱和食盐水溶液洗涤有机层;萃取剂为二氯甲烷;采用无水硫酸镁进行干燥,柱色谱四为sio2/二氯甲烷/正己烷2:1。
步骤6、磁性介孔分子印迹聚合物fe3o4@mtio2@mip的制备;
具体为,将0.21g的4-乙烯基吡啶与tbbpa溶于50ml无水甲苯中,在氮气的保护下,机械搅拌12-24h;加入50mg的raft试剂修饰的磁性介孔二氧化钛载体fe3o4@mtio2-cpdb,将其分散于上述溶液中;之后加入1.189g的乙二醇二甲基丙烯酸酯和20mg的偶氮二异丁腈,在氮气气氛下,进行聚合反应,得到反应产物;并采用乙醇对反应产物进行洗涤,真空干燥得到所述的磁性介孔分子印迹聚合物;其中反应温度为60-70℃,反应时间为16-32h。
步骤7、去除模板分子
具体为,将干燥后的磁性介孔分子印迹聚合物包裹在滤纸中,并将滤纸包裹的干燥后的磁性表面分子印迹聚合物置于索氏提取器中,经甲醇-乙酸溶液抽提,得到没有模板分子的磁性表面分子印迹聚合物fe3o4@mtio2@mip;其中甲醇-乙酸溶液采用甲醇与乙酸按体积比为9:1的比例配制得到。
如附图2所示,附图2给出了实施例1中制备得到的磁性表面分子印迹聚合物的透射电子显微图像;从附图2中可以看出,实施例1中制备的磁性表面分子印迹聚合物表现出球形结构且分散性良好并具有明显的多层核壳结构;最内层的核芯为fe3o4纳米颗粒,直径约为200-300nm;紧贴fe3o4纳米颗粒的一层为均匀包覆的sio2层,其厚度约为60-65nm;紧接着一层为通过动力学控制方法均匀包覆的介孔tio2层,厚度约为22-30nm;最外面也是最薄的一层为聚合物层;聚合物层位于载体表面有利于模板结合和去除过程中的快速传质。
如附图3所示,附图3中给出了实施例1中制备的fe3o4磁性纳米颗粒(a)、包覆二氧化硅后的磁性纳米微球fe3o4@sio2(b)、磁性介孔二氧化钛纳米微球fe3o4@mtio2(c)、叠氮功能化的磁性纳米介孔载体fe3o4@mtio2-n3(d)、raft试剂修饰后的磁性介孔二氧化钛载体fe3o4@mtio2-cpdb(e)以及磁性表面分子印迹聚合物fe3o4@mtio2@mip(f)的红外光谱图;
从附图3(a)中可以看出,450和570cm-1处的吸收峰可归属于fe-o官能团的振动吸收峰,1390cm-1附近的吸收峰可归属于fe3o4的制备过程中所加入的柠檬酸钠中的羧酸根的特征吸收峰。
从附图3(b)中可以看出在1091cm-1处的新吸收峰的出现,对应于si-o-si的伸缩振动特征吸收峰,805cm-1附近的吸收峰是由si-o-si的对称伸缩振动引起的,表明sio2被成功包覆。
从附图3(c)中可以看出,400-700cm-1处的特征吸收峰属于ti-o-ti和ti-o的伸缩振动吸收峰,表明tio2层被成功地包覆在sio2表面。
从附图3(d)中可以看出在2100cm-1处的吸收峰,归属于叠氮基的特征吸收峰,表明叠氮基被成功修饰在fe3o4@mtio2表面。
从附图3(e)中可以看出,经raft试剂修饰后,出现了位于1166cm-1处的c=s的特征峰,表明cpdb通过点击化学被成功修饰。
从附图3(f)中可以看出,在1457cm-1处观察到来自吡啶环中c=c的伸缩振动特征峰,以及1741cm-1和1647cm-1处乙二醇二甲基丙烯酸酯的c=o和c-o的伸缩振动特征峰证实mip层被成功地接枝在fe3o4@mtio2的表面上。
如附图4所示,附图4中给出了实施例1中制得的fe3o4磁性纳米颗粒(a)、包覆二氧化硅后的磁性纳米微球fe3o4@sio2(b)、磁性介孔二氧化钛纳米微球fe3o4@mtio2(c)以及磁性表面分子印迹聚合物fe3o4@mtio2@mip(f)的x射线衍射谱图;
从附图4(a)中可以看出fe3o4纳米粒子衍射谱图中出现了fe3o4纳米粒子位于30.0°,35.5°,43.2°,53.8°,57.2°以及62.6°处的衍射峰,分别对应于(220),(311),(400),(422),(511)以及(440)晶面。
从附图4(b)中可以看出包覆了sio2层之后,衍射谱图并无明显变化,说明所包覆的sio2层为无定型结构。
从附图4(c)中可以看出,新增加的特征衍射峰与锐钛矿地特征衍射峰匹配,表明tio2被成功引入。
从附图4(d)中可以看出,衍射谱图与附图4(c)没有明显差异,说明在制备磁性表面分子印迹聚合物fe3o4@mtio2@mip的过程中不影响磁铁矿核和tio2的晶体结构。
如附图5所示,附图5中给出了实施例1中制得的fe3o4磁性纳米颗粒(a)、包覆二氧化硅后的磁性纳米微球fe3o4@sio2(b)、磁性介孔二氧化钛纳米微球fe3o4@mtio2(c)以及磁性表面分子印迹聚合物fe3o4@mtio2@mip(d)的磁滞回线图;
从附图5(a)中可以看出所制备的纯fe3o4的比饱和磁化强度为40.5emu/g。
从附图5(b)中可以看出包覆了sio2之后,由于非磁性成分的引入,比饱和磁化强度降低为31.7emu/g。
从附图5(c)中可以看出介孔tio2的引入使得比饱和磁化强度又降低为28.7emu/g,但是随着壳层厚度的增加,样品仍然保持着超顺磁性。
从附图5(d)中可以看出比饱和磁化强度值在包覆了印迹聚合物层之后还会有所降低,但是也间接地说明了印迹聚合物层被成功地引入到介孔tio2载体表面。虽然经包覆印迹聚合物之后比饱和磁化强度值降低至23.6emu/g,但仍然能够通过施加外部磁场容易地分离回收。
从附图5中可知,所有样品都表现出超顺磁性,随着壳层厚度的增加,磁化强度会有所降低是由于sio2,tio2和mip层的包覆引起的,但它们仍然具有较高的比饱和磁化强度,可以在外部磁场的作用下快速完全分离。
本发明所述的一种磁性表面分子印迹聚合物,采用磁性介孔二氧化钛纳米微球fe3o4@mtio2作为印迹聚合的载体制备磁性表面分子印迹聚合物;制备的印迹材料能够在短时间内特异性识别tbbpa。磁核的存在使得印迹材料可以通过外加磁场快速分离,介孔结构的高比表面积增加了印迹材料的结合位点;印迹层的表层分布也使得印迹位点容易接近,达到迅速识别模板分子的效果。良好的可分离回收性和选择性,使得该印记聚合物有望在实际应用中实现对复杂环境中tbbpa的识别、检测。
实施例2
本实施例与实施例1步骤大体相同,不同之处在于步骤6中4-乙烯基吡啶与乙二醇二甲基丙烯酸酯的比例。
具体的,步骤6中,将0.21g4-乙烯基吡啶与tbbpa溶于50ml无水甲苯中,在氮气的保护下,机械搅拌12-24h;加入50mgraft试剂修饰的磁性介孔二氧化钛载体fe3o4@mtio2-cpdb,将其分散于上述溶液中;之后加入0.369g乙二醇二甲基丙烯酸酯和20mg偶氮二异丁腈,在氮气气氛下进行聚合反应,得到反应产物;采用乙醇对反应产物进行洗涤,真空干燥得到所述的磁性表面分子印迹聚合物fe3o4@mtio2@mip;其中聚合反应温度为60-70℃,反应时间为16-32h。在不同比例下获得的印迹聚合物,对tbbpa表现出不同的结合能力,其中功能单体与交联剂比例为1:1时印迹因子为1.71。
实施例3
本实施例与实施例1步骤大体相同,不同之处在于步骤6中4-乙烯基吡啶与乙二醇二甲基丙烯酸酯的比例。
具体的,步骤6中,将0.21g4-乙烯基吡啶与tbbpa溶于50ml无水甲苯中,在氮气的保护下,机械搅拌12-24h;加入50mg的raft试剂修饰的磁性介孔二氧化钛载体fe3o4@mtio2-cpdb,将其分散于上述溶液中;之后加入1.189g乙二醇二甲基丙烯酸酯和20mg偶氮二异丁腈,在氮气气氛下,进行聚合反应,得到反应产物;采用乙醇对反应产物进行洗涤,真空干燥得到所述的磁性表面分子印迹聚合物fe3o4@mtio2@mip;其中聚合反应温度为60-70℃,反应时间为16-32h。在不同比例下获得的印迹聚合物,对tbbpa表现出不同的结合能力,其中功能单体与交联剂比例为1:3时印迹因子为2.00。
实施例4
本实施例与实施例1步骤大体相同,不同之处在于步骤6中4-乙烯基吡啶与乙二醇二甲基丙烯酸酯的比例。
具体的,步骤6中,将0.21g4-乙烯基吡啶与tbbpa溶于50ml无水甲苯中,在氮气的保护下,机械搅拌12-24h;加入50mgraft试剂修饰的磁性介孔二氧化钛载体fe3o4@mtio2-cpdb,将其分散于上述溶液中;之后加入1.979g乙二醇二甲基丙烯酸酯和20mg偶氮二异丁腈,在氮气气氛下,进行聚合反应,得到反应产物;采用乙醇对反应产物进行洗涤,真空干燥得到所述的磁性表面分子印迹聚合物fe3o4@mtio2@mip;其中反应温度为60-70℃,反应时间为16-32h。在不同比例下获得的印迹聚合物,对tbbpa表现出不同的结合能力,其中功能单体与交联剂比例为1:5时印迹因子为1.76。
对比例1
对比例1与实施例1步骤大体相同,不同之处在于步骤6中聚合的时间。
具体的,步骤6中,将0.21g4-乙烯基吡啶与tbbpa溶于50ml无水甲苯中,在氮气的保护下,机械搅拌12-24h;加入50mgraft试剂修饰的磁性介孔二氧化钛载体fe3o4@mtio2-cpdb,将其分散于上述溶液中;之后加入1.189g乙二醇二甲基丙烯酸酯和20mg偶氮二异丁腈,在氮气气氛下,进行聚合反应,得到反应产物;采用乙醇对反应产物进行洗涤,真空干燥得到所述的磁性表面分子印迹聚合物fe3o4@mtio2@mip;其中反应温度为60-70℃,反应时间为12h。对对比例1制备得到的磁性表面分子印迹聚合物fe3o4@mtio2@mip通过吸附实验进行性能评估,发现所得到的印迹聚合物对18.5μmol/ltbbpa的去除效率8.0%。可能是由于聚合时间太短,功能单体与交联剂之间的聚合程度不够,所形成的的印迹腔刚性较低,而且识别位点有限,造成对模板分子较低的去除率。
对比例2
对比例2与实施例1步骤大体相同,不同之处在于步骤6中聚合的时间。
具体的,步骤6中,将0.21g4-乙烯基吡啶与tbbpa溶于50ml无水甲苯中,在氮气的保护下,机械搅拌12-24h;加入50mgraft试剂修饰的磁性介孔二氧化钛载体fe3o4@mtio2-cpdb,将其分散于上述溶液中;之后加入0.369乙二醇二甲基丙烯酸酯和20mg偶氮二异丁腈,在氮气气氛下,进行聚合反应,得到反应产物;采用乙醇对反应产物进行洗涤,真空干燥得到所述的磁性表面分子印迹聚合物fe3o4@mtio2@mip;其中反应温度为60-70℃,反应时间为48h。对对比例2制备得到的磁性表面分子印迹聚合物fe3o4@mtio2@mip通过吸附实验进行性能评估,发现其对18.5μmol/ltbbpa的去除效率为11.8%。可能是聚合时间过长,会造成介孔载体孔道结构的堵塞,使得载体高比表面积的性质没法充分发挥,去除率较低。
在实际应用中,本发明所述的一种磁性表面分子印迹聚合物能够选择性能和可回收的结果表明有希望在复杂环境中对tbbpa进行识别和检测。
等温吸附试验
在等温吸附试验中,取八组磁性表面分子印迹聚合物和八组tbbpa的甲醇-水溶液,其中每组磁性表面分子印迹聚合物的质量为10mg,每组tbbpa的甲醇-水溶液的体积为10ml,其中八组tbbpa的甲醇-水溶液中tbbpa的浓度分别为18.5μmol/l、37.0μmol/l、55.5μmol/l、74.0μmol/l、92.5μmol/l、111.0μmol/l、148.0μmol/l及185.0μmol/l,甲醇和水的体积比为1:1,在上述八组tbbpa的甲醇-水溶液分别加入一组磁性表面分子印迹聚合物,均置于30-35℃的恒温振荡器中震荡过夜,磁分离后,通过液相色谱测定上述八组含tbbpa的甲醇-水溶液的上清液中的tbbpa的浓度依次为12.61μmol/l、26.84μmol/l、39.65μmol/l、54.90μmol/l、69.99μmol/l、85.15μmol/l、119.04μmol/l及148.92μmol/l。
取八组非印迹聚合物和八组tbbpa的甲醇-水溶液,其中每组非印迹聚合物的质量为10mg,每组tbbpa的甲醇-水溶液的体积为10ml,其中八组tbbpa的甲醇-水溶液中tbbpa的浓度分别为18.5μmol/l、37.0μmol/l、55.5μmol/l、74.0μmol/l、92.5μmol/l、111.0μmol/l、148.0μmol/l及185.0μmol/l,甲醇和水的体积比为1:1,在上述八组tbbpa的甲醇-水溶液分别加入一组非印迹聚合物,均置于30-35℃的恒温振荡器中震荡过夜,磁分离后,通过液相色谱测定上述八组tbbpa的甲醇-水溶液的上清液中的tbbpa的浓度依次为16.98μmol/l、33.79μmol/l、50.75μmol/l、68.68μmol/l、86.49μmol/l、103.31μmol/l、138.66μmol/l、172.85μmol/l;由此可知,本发明所述的磁性表面分子印迹聚合物对tbbpa的吸附量在相同条件下明显高于非印迹聚合物,本发明所述的磁性表面分子印迹聚合物相比于非印迹聚合物对tbbpa具有更好的亲和力。
吸附动力学试验
在吸附动力学试验中,将10mg的磁性表面分子印迹聚合物加入到10ml的tbbpa的甲醇-水溶液,将10mg的非印迹聚合物加入到10ml的tbbpa的甲醇-水溶液;其中,tbbpa的甲醇-水溶液中tbbpa的浓度为18.5μmol/l,甲醇和水的体积比为1:1;分别置于30-35℃的恒温振荡器中震荡一定时间,磁分离后,通过液相色谱测定上清液中的tbbpa的浓度;其中,含有磁性表面分子印迹聚合物的tbbpa的甲醇-水溶液的tbbpa的浓度在5min、10min、15min、20min、30min、60min、100min时分别为10.22μmol/l、9.31μmol/l、8.12μmol/l、6.90μmol/l、6.96μmol/l、6.63μmol/l、6.57μmol/l,含有非印迹聚合物的tbbpa的甲醇-水溶液的tbbpa的浓度在5min、10min、15min、20min、30min、60min、100min时分别为13.52μmol/l、12.77μmol/l、12.37μmol/l、12.14μmol/l、11.88μmol/l、11.68μmol/l、11.53μmol/l;由此可知,本发明所述的磁性表面分子印迹聚合物由于识别位点位于载体表面进而可以在短时间内达到吸附平衡,并且相同条件下吸附量远高于在非印迹聚合物。
选择性吸附试验
在选择性吸附试验中,采用选择tbbpa的结构类似物,例如:双酚a,4,4'-二羟基联苯及4-叔丁基苯酚来研究印迹聚合物的选择性;
具体的,将10mg的磁性表面分子印迹聚合物加入到10ml的tbbpa及其结构类似物的甲醇-水溶液,将10mg的非印迹聚合物加入到10ml的tbbpa及其结构类似物的甲醇-水溶液;其中,tbbpa及其结构类似物的甲醇-水溶液中tbbpa及其结构类似物的浓度为18.5μmol/l,甲醇和水的体积比为1:1;吸附平衡后,通过测定上清液中的tbbpa及其结构类似物的浓度;其中,加入磁性表面分子印迹聚合物的甲醇-水溶液中tbbpa、双酚a、4,4'-二羟基联苯及4-叔丁基苯酚的浓度分别为8.58μmol/l、15.58μmol/l、16.47μmol/l、17.54μmol/l,加入非印迹聚合物的甲醇-水溶液中tbbpa、双酚a、4,4'-二羟基联苯及4-叔丁基苯酚的浓度分别为11.95μmol/l、16.98μmol/l、17.72μmol/l、17.69μmol/l;由此可知,印迹聚合物对tbbpa的去除效率显着高于其他三种物质,这证明了tbbpa的印迹材料的特异性识别。原因是合成的fe3o4@mtio2@mip表面产生特定的识别腔完全匹配于tbbpa的形状,大小和空间排布,而bip,bp和bpa不与识别位点互补,因此去除效率低。
竞争性吸附试验
在竞争性吸附试验中,将10mg的磁性表面分子印迹聚合物加入到10ml的tbbpa及其结构类似物的甲醇-水溶液。其中,tbbpa及其结构类似物的甲醇-水溶液中tbbpa、双酚a、4,4'-二羟基联苯及4-叔丁基苯酚的浓度均为18.5μmol/l,甲醇和水的体积比为1:1;吸附平衡后,通过测定上清液中的tbbpa及其结构类似物的浓度;其中,含有磁性表面分子印迹聚合物的tbbpa的甲醇-水溶液中的tbbpa、双酚a、4,4'-二羟基联苯及4-叔丁基苯酚的浓度分别为11.06μmol/l、18.00μmol/l、17.97μmol/l及17.23μmol/l;由此可知,fe3o4@mtio2@mip仅对tbbpa表现出更好的去除效率。相反,由于其对tbbpa的高选择性,其他三种物质的去除效率非常小,这表明本发明所述的磁性表面分子印迹聚合物能够在复杂环境中特异性识别tbbpa。
- 还没有人留言评论。精彩留言会获得点赞!