一种化合物及其制备方法和应用与流程
本发明涉及医药技术领域,尤其涉及一种化合物及其制备方法和应用。
背景技术:
心脑血管疾病是心脏血管和脑血管疾病的统称,泛指由于高脂血症、血液粘稠、动脉粥样硬化、高血压等导致的心脏、大脑及全身组织发生的缺血性或出血性疾病。心脑血管疾病患病率高、致残率高、死亡率高,已经成为全世界范围内严重威胁人类健康的头号杀手。随着经济发展,人们生活水平逐步提高,生活节奏不断加快,使心脑血管疾病的发病率不断攀升,而预防和治疗心脑血管疾病的药物需求也随之增加,在国际药品市场上占有举足轻重的地位,因此研究和开发心脑血管疾病用药成为当今最热门的研究领域之一。
技术实现要素:
有鉴于此,本发明提供一种从银杏叶中分离制备出一种抗心脑血管疾病的化合物及其制法和应用。
本发明提出了一种具有式i所示结构的化合物或者其药学上可接受的盐、溶剂化物、异构体、前药分子或代谢物,该化合物具有如下结构:
本发明还提出了式i所示化合物的制备方法,其特征在于,该方法包括:
a)对银杏叶进行提取,得到清膏;
b)将得到的清膏进行分离,得到式i所示化合物。
其中,本发明对银杏叶提取的方法没有特殊限制,本领域公知的用于溶剂提取中草药成分的方法均可,本发明优选采用一定浓度的乙醇水溶液提取。
具体的,银杏叶与乙醇的用量比(以质量计)优选为1:(6~10),在本发明的某些具体实施例中,所述用量比为1:8;所述提取的次数优选为1~3次,在本发明的某些具体实施例中,所述提取的次数为2次;所述提取的时间优选为0.5~3小时,更优选为1.5~2.5小时;本发明对采用的乙醇并无特殊限定,可以为普通市售乙醇,优选为百分含量50%~95%的乙醇,更优选为百分含量60%的乙醇。
具体地,所述步骤b)包括:
将得到的清膏通过大孔吸附树脂柱色谱、反相柱色谱、凝胶柱色谱和制备型液相色谱进行分离,得到式i所示化合物。
具体地,所述大孔吸附树脂柱色谱中的大孔吸附树脂选自d101型大孔吸附树脂、hp-20型大孔吸附树脂、hpd-450型大孔吸附树脂、hpd-950型大孔吸附树脂和ab-8型大孔吸附树脂中的一种或几种。其中,所述洗脱液优选为乙醇-水溶液;乙醇与水的体积比为(x):(100-x)的溶液,其中,0≤x≤95,更优选为依次采用水,(20~35)%乙醇-水溶液进行洗脱,收集(20~35)%乙醇-水溶液洗脱部位,得到大孔树脂分离后的粗品。
具体地,所述反相柱色谱选自动态轴向压缩色谱、高效液相色谱或中低压制备色谱;采用的洗脱液为甲醇-水溶液或者乙腈-水溶液。所述洗脱剂优选为乙腈-水溶液,且为了能够更好分离杂质和所需化合物,本发明优选采用梯度洗脱,所述梯度洗脱程序为0~70min,20~80%乙腈-水溶液,流速100ml/min,收集25~35min洗脱液,得反相柱色谱分离后的粗品。
具体地,所述凝胶柱色谱中的凝胶选自sephadexlh-20、sephadexg15或sephadexg50。
具体地,所述制备型液相色谱采用的流动相为甲醇-水溶液或者乙腈-水溶液。其中,所用流动相优选为乙腈-水溶液,优选的为(15~45)%乙腈-水溶液,最优选为(20~40)%乙腈-水溶液;所述流速优选为3~100ml/min,最优选为5~30ml/min;检测波长为340nm,得到式i所示化合物。
更具体地,本发明式i所示化合物的制备方法包括:
将银杏叶与50%~95%的乙醇按1:(6~10)混合,提取1~3次,每次提取0.5~3小时,将提取液浓缩得到清膏;
将得到的清膏通过大孔吸附树脂柱,依次采用水、(20~35)%乙醇-水溶液进行洗脱,收集(20~35)%乙醇-水溶液洗脱部位,得到大孔树脂分离后的粗品;
将大孔树脂分离后的粗品通过反相柱色谱分离,其采用梯度洗脱,所述梯度洗脱程序为0~70min,20~80%乙腈-水溶液,收集25~35min洗脱液,得到反相柱色谱分离后的粗品;
将反相柱色谱分离后得到的粗品通过sephadexlh-20凝胶柱色谱分离,洗脱液为甲醇,得到凝胶分离后的粗品;
将凝胶分离后的粗品通过制备型液相色谱分离,所用流动相为(15~45)%乙腈-水溶液,所述流速为3~100ml/min。
优选地,本发明式i所示化合物的制备方法包括:
干燥的银杏叶10kg,粉碎过筛后加入10倍量的60%乙醇,提取3次,每次1.5h,过滤,过滤浓缩、回收乙醇得到清膏;
取上述清膏加入水使溶解,室温静置,取上清液经hp-20大孔吸附树脂柱分离,依次采用水,35%乙醇-水溶液进行洗脱,收集35%乙醇-水溶液的洗脱部位;
将35%35%乙醇-水溶液的洗脱部位,经反相c18动态轴向压缩柱色谱分离,以0~70min,20~80%乙腈-水溶液进行梯度洗脱,流速100ml/min,收集25~35min洗脱液;
取前述洗脱液,经sephadexlh-20柱色谱分离,甲醇洗脱,进行纯化。
将纯化后的样品,经制备型hplc分离,以20%乙腈-水溶液为流动相,检测波长为340nm,流速15ml/min。
本发明还提出了式i所示化合物或者其药学上可接受的盐、溶剂化物、异构体、前药分子或代谢物在制备抗心脑血管疾病药物中的应用。
本发明还提出了一种抗心脑血管疾病药物,其特征在于,包括权利要求1所述的化合物或者其药学上可接受的盐、溶剂化物、异构体、前药分子或代谢物,和药学上可接受的辅料。其中,该辅料可根据剂型和实际情况进行恰当选择,例如常用的辅料有淀粉、低取代羟丙基纤维素、微粉硅胶、硬脂酸镁、淀粉浆、蔗糖、糊精、羧甲基淀粉钠、滑石粉、聚山梨酯、聚乙二醇、注射用大豆磷脂和注射用甘油等;利用本发明得到的式i所示化合物,制备所需药物的各种剂型时,可以按照药剂学领域的常规生产方法制备。如将该提取物与一种或多种载体混合,然后制成相应的剂型。
具体地,所述抗心脑血管疾病药物的剂型为口服给药剂型、注射给药剂型或外用给药制剂。
进一步地,所述抗心脑血管疾病药物可以是药剂学上所说的任何一种剂型,包括片剂、胶囊剂、软胶囊、凝胶剂、口服剂、混悬剂、冲剂、贴剂、软膏、丸剂、散剂、注射剂、输液剂、冻干注射剂、静脉乳剂、脂质体注射剂、栓剂、缓释制剂或控释制剂。
本发明还提出了化合物阿魏酰基槲皮素或山奈酚,其中,阿魏酰基槲皮素或山奈酚,是指槲皮素或山奈酚或它们的苷类成分中的至少一羟基氢原子被阿魏酰基取代。可选地,它们的苷类成分被取代的羟基为6位oh。如:进一步可选地为quercetin3-o-(6-trans-feruloyl)-β-glucopyranosyl-(1→2)-β-glucopyranoside。
本发明还提出了槲皮素、山奈酚、阿魏酰基槲皮素或山奈酚,或其(槲皮素、山奈酚、阿魏酰基槲皮素或山奈酚)药学上可接受的盐、溶剂化物、异构体、前药分子或代谢物在制备抗心脑血管疾病药物中的应用。
本发明通过对银杏叶进行提取,得到清膏;然后将得到的清膏进行分离,选择特定出峰时间的化合物,即得到式i所示化合物,且通过细胞实验发现,该化合物对h2o2所致h9c2细胞损伤和垂体后叶素致大鼠实验性心肌缺血具有保护作用,显著降低ldh水平、显著升高血浆中ck水平和显著降低血浆mda水平,可开发成抗心脑血管疾病药物,具有良好的应用前景。
附图说明
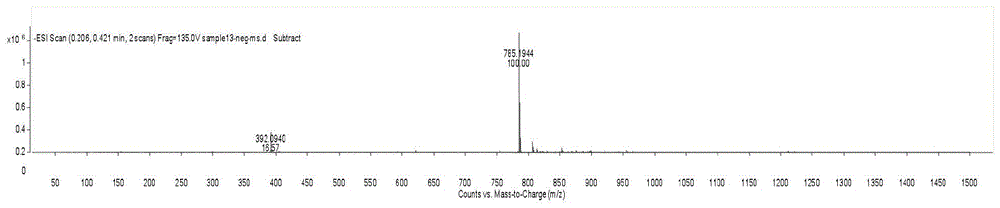
图1为本发明实施例1制得的式i结构的化合物的esi-ms谱图
图2为本发明实施例1制得的式i结构的化合物的1h-nmr谱图;
图3为本发明实施例1制得的式i结构的化合物的13c-nmr谱图;
图4为本发明实施例1制得的式i结构的化合物的hsqc谱图;
图5为本发明实施例1制得的式i结构的化合物的1h-1hcosy谱图;
图6为本发明实施例1制得的式i结构的化合物的hmbc谱图;
图7为本发明实施例1制得的式i结构的化合物的noesy相关。
具体实施方式
以下结合附图和实施例,对本发明的具体实施方式进行更加详细的说明,以便能够更好地理解本发明的方案以及其各个方面的优点。然而,以下描述的具体实施方式和实施例仅是说明的目的,而不是对本发明的限制。
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
其次,需要注意的是,本发明所涉及的未注明的浓度均为体积百分含量(v/v)。其它除非另外说明,否则所有的百分数、比率、比例或份数按重量计。另,本发明如未注明具体条件者,均按照常规条件或制造商建议的条件进行,所用原料药或辅料,以及所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
除非另行定义,文中所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明。
为了进一步说明本发明,下面结合实施例对本发明提供的化合物及其制备方法和应用进行详细描述。
实施例1式ⅰ所示结构的化合物的制备
1)干燥的银杏叶10kg,粉碎过筛后加入10倍量的60%乙醇,提取3次,每次1.5h,过滤,过滤浓缩、回收乙醇得到清膏;
2)取步骤1)的清膏加入纯化水使溶解,室温静置,取上清液经hp-20大孔吸附树脂柱分离,依次采用水、35%乙醇-水溶液进行洗脱,每种洗脱液洗脱4个柱体积(下同),收集35%乙醇-水溶液洗脱部位;
3)取步骤2)的35%乙醇-水溶液洗脱部位,经反相c18动态轴向压缩柱色谱分离,以0~70min,20~80%乙腈-水溶液进行梯度洗脱,流速100ml/min,收集25~35min洗脱液;
4)取步骤3)的洗脱液,经sephadexlh-20柱色谱分离,甲醇洗脱,进行纯化;
5)取步骤4)纯化后的样品,经制备型hplc分离,以20%乙腈-水溶液为流动相,检测波长为340nm,流速15ml/min,分离所得溶液干燥,得到本发明所述的式ⅰ结构的化合物10mg,纯度为98.5%。
上述方法得到的化合物为黄色无定形粉末。盐酸-镁粉反应呈阳性,molish反应呈阳性,表明其可能为黄酮苷类化合物。高分辨质谱hr-esi-ms表征结果m/z785.1944[m-h]—(计算值为785.1935),表明化合物分子量为786。结合元素分析及13c-nmr谱推断该化合物分子式为c37h38o19。
如图1~图7所示,图1为本发明实施例1制得的式i结构的化合物的esi-ms谱图,图2为本发明实施例1制得的式i结构的化合物的1h-nmr谱图,图3为本发明实施例1制得的式i结构的化合物的13c-nmr谱图,图4为本发明实施例1制得的式i结构的化合物的hsqc谱图,图5为本发明实施例1制得的式i结构的化合物的1h-1hcosy谱图,图6为本发明实施例1制得的式i结构的化合物的hmbc谱图,图7为本发明实施例1制得的式i结构的化合物的noesy谱图。
本发明实施例1化合物的1h-nmr(dmso-d6,400mhz)谱(见图2),δh12.67(1h,s,5-oh)为黄酮类化合物5位活泼羟基特征信号;低场区芳香质子δh6.18(1h,s,h-6)和6.33(1h,s,h-8)为a环间位耦合的两个芳香质子信号;δh6.90(1h,d,j=8.4hz,h-5′)、7.26(1h,d,j=8.4,2.0hz,h-6′)和7.38(h,d,j=2.0hz,h-2′),δh6.72(1h,d,j=8.2hz,h-8"”)、6.99(1h,dd,j=8.2,1.7hz,h-9"”)和7.21(1h,d,j=1.7hz,h-5"”)两组abx系统;δh6.35(1h,d,j=15.9hz,h-2"”)和7.45(1h,d,j=15.9hz,h-3"”)一组反式双键。δh4.29(1h,d,j=7.8hz,h-1"')为葡萄糖端基质子信号,据其偶合常数可知葡萄糖苷键为β-构型。δh5.52(1h,s,h-1")为鼠李糖端基质子信号,δh0.92(3h,d,j=6.2hz,h-6")为鼠李糖基上的甲基质子特征信号。δh3.75(3h,s,6"”-och3)为甲氧基质子信号。
13c-nmr(dmso-d6,400mhz)谱中(见图3),共给出37个碳信号,低场24个sp2杂化碳,其中15个为黄酮母核碳信号,δc134.7(c-3)为典型的黄酮醇3位碳信号;剩余9个碳信号,结合氢谱推断,1个羰基碳信号,2个双键碳信号,6个苯环碳信号,为苯丙酰基片段。高场区12个糖上的碳信号,一个甲氧基碳信号。
通过hsqc图谱(见图4),具有相关关系的碳信号与氢信号进行归属(见表1),在hmbc图谱(见图6),δh6.90(1h,d,j=8.4hz,h-5′)和7.38(h,d,j=2.0hz,h-2′)与158.9(c-2)相关,提示该黄酮母核为槲皮素;鼠李糖端基质子δh5.52(1h,s,h-1")与黄酮母核3位碳δc134.7(c-3)远程相关,提示苷元的3位与鼠李糖的1位相连;葡萄糖端基质子δh4.29(1h,d,j=7.8hz,h-1"')与鼠李糖2碳信号δc82.2(c-2")有远程相关,表明葡萄糖1位与鼠李糖2位相连;1组abx系统δh6.72(1h,d,j=8.2hz,h-8"”)、6.99(1h,dd,j=8.2,1.7hz,h-9"”)和7.21(1h,d,j=1.7hz,h-5"”)与双键碳δc145.5(c-3"”)相关,甲氧基质子δh3.75(3h,s,6"”-och3)与δc148.3(c-6"”)远程相关,结合noesy谱,推断该苯丙酰基片段为阿魏酰基;δh4.04(1h,d,j=11.8hz,h-6"'a)、4.22(1h,dd,j=11.8,3.8hz,h-6"'b)与δc166.9(c-1"”)远程相关,提示阿魏酰基与葡萄糖6位相连。
综合以上数据分析,最终确定本发明化合物的结构为quercetin3-o-(6-trans-feruloyl)-β-glucopyranosyl-(1→2)-α-rhamnopyranoside,中文名为槲皮素-3-o-6-阿魏酰基葡萄糖-(1→2)-鼠李糖苷,结构如式i所示,为一个新的黄酮类化合物。所有的碳氢信号归属见表1,表1为式i所示化合物的各个碳和氢的归属。
表1化合物的核磁数据(dmso-d6,1h-nmr400mhz,13c-nmr100mhz)
通过对检测结果分析可知,本发明得到的化合物的结构如式i所示。
实施例2式ⅰ所示结构的化合物的制备
1)干燥的银杏叶10kg,粉碎过筛后加入8倍量的70%乙醇,提取2次,每次2h,过滤,过滤浓缩、回收乙醇得到清膏;
2)取步骤1)的清膏加入纯化水使溶解,室温静置,取上清液经d101大孔吸附树脂柱分离,依次采用水、20%乙醇-水溶液进行洗脱,收集20%乙醇-水溶液洗脱部位;
3)取步骤2)的20%乙醇-水溶液洗脱部位,经反相c18动态轴向压缩柱色谱分离,以0~70min,20~80%乙腈-水溶液进行梯度洗脱,流速100ml/min,收集25~35min洗脱液;
4)取步骤3)的洗脱液,经sephadexlh-20柱色谱分离,甲醇洗脱,进行纯化。
5)取步骤4)纯化后的样品,经制备型hplc分离,以40%乙腈-水溶液为流动相,检测波长为340nm,流速15ml/min,分离所得溶液干燥,得到本发明所述的式ⅰ结构所示的化合物9mg,纯度为98.6%。
按实施例1的方法通过对检测结果分析可知,本发明得到的化合物结构如式i所示。
实施例3式ⅰ所示结构的化合物的制备
1)干燥的银杏叶10kg,粉碎过筛后加入6倍量的70%乙醇,提取2次,每次2h,过滤,过滤浓缩、回收乙醇得到清膏;
2)取步骤1)的清膏加入纯化水使溶解,室温静置,取上清液经d101大孔吸附树脂柱分离,依次采用水、20%乙醇-水溶液进行洗脱,收集20%乙醇-水溶液洗脱部位;
3)取步骤2)的20%乙醇-水溶液洗脱部位,经反相c18动态轴向压缩柱色谱分离,以0~70min,20~80%乙腈-水溶液进行梯度洗脱,流速100ml/min,收集25~35min洗脱液;
4)取步骤3)的洗脱液,经sephadexlh-20柱色谱分离,甲醇洗脱,进行纯化。
5)取步骤4)纯化后的样品,经制备型hplc分离,以45%乙腈-水溶液为流动相,检测波长为340nm,流速15ml/min,分离所得溶液干燥,得到本发明所述的式ⅰ结构的化合物9mg,纯度为98.5%。
按实施例1的方法通过对检测结果分析可知,本发明得到的化合物结构如式i所示。
实施例4本发明化合物对h2o2所致h9c2细胞损伤的保护作用研究1.材料
1.1药物:式i所示化合物;
阳性对照药1:抗坏血酸;
对照药1:
quercetin3-o-(6-trans-feruloyl)-β-glucopyranosyl-(1→2)-β-glucopyranoside;
对照药2:槲皮素
对照药3:山奈酚
1.2细胞模型:h9c2大鼠心肌细胞株,来源于中国科学院上海细胞库;培养条件:dmem+10%胎牛血清,37℃,5%co2;
2.实验方法与步骤
(1)药液的配制:将药物以dmso溶解,制得1mol/l的储备液。临用时分别稀释成高、中、低浓度为50、25、12.5μmol/l的药液。
(2)各对照药液的配制:将抗坏血酸、quercetin3-o-(6-trans-feruloyl)-β-glucopyranosyl-(1→2)-β-glucopyranoside、槲皮素、山奈酚分别用dmso溶解,制成浓度为25μmol/l的对照药液(分别对应于阳性对照组1、对照组1、对照组2、对照组3)。
(3)实验方法:将细胞以1×105个/ml的浓度均匀接种于96孔板,每孔100μl,种板后于37℃,5%co2细胞培养箱中培养24小时。培养24小时后,取出96孔板,吸弃上清液,按照试验要求随机分为空白组、模型组、对照组和给药组。
空白组:每孔加入100μl无血清的dmem培养基;
模型组、对照组和给药组分别给予100μl含浓度为200μmol/lh2o2的无血清培养基;氧化损伤3h后,空白组和模型组分别给予100μl完全培养基,对照组均给予100μl浓度为25μmol/l的对照药,给药组给予100μl浓度分别为50μmol/l、25μmol/l、12.5μmol/l的高、中、低剂量组的式ⅰ所示化合物药液。复氧3h后,每孔加入20μl的mts溶液培养4h,于490nm下测定吸光度(a)值。根据公式计算细胞保护率。
细胞保护率(%)=(a给药-a模型)/(a空白-a模型)×100%。
3.实验结果
实验结果表明,给药组和各对照组均可以显著提高损伤细胞的存活率,显示出较强的细胞保护活性。数据结果如表1。
表1式i化合物对h2o2所致h9c2细胞损伤的保护影响(x±s,n=6)
与空白组比较,##p<0.01,与模型组比较,**p<0.01,*p<0.05
4.结论
h2o2是反应性氧代谢产生的原发性化学物质,可以进一步产生其他更多的反应性氧化剂,可促进自由基生成。本发明化合物对h2o2所致h9c2细胞损伤具有显著的保护作用,显示出较强的抗氧化活性,且随着药物浓度的上升,抗氧化强度越大,对细胞损伤的保护作用也随之增加。
对照组2与对照组3相比,槲皮素组优于山奈酚组,提示黄酮母核临二酚羟基抗氧化活性更强;
对照组1和中剂量组均优于对照组2,提示带阿魏酰基的槲皮素糖苷抗氧化作用优于槲皮素苷元;
对照组1与中剂量相比,中剂量优于对照组1,提示6-阿魏酰基葡萄糖-(1→2)-鼠李糖苷结构优于6-阿魏酰基葡萄糖-(1→2)-葡萄糖苷。
实施例5本发明化合物对垂体后叶素致大鼠实验性心肌缺血模型的作用研究
1.材料
1.1实验动物spf级sd大鼠60只,雌雄各半,体重150~180g,由浙江省实验动物中心提供,合格证号:scxk(浙)20130033。
1.2药品及试剂盐酸维拉帕米片,40mg/片,上海信谊药厂有限公司,批号:131101b;垂体后叶注射液,6单位/2ml,上海第一生化药品有限公司,批号:130501;sod,mda试剂盒均购自于京华英生物技术研究所,批号分别为:20130101m,20130410;ldh、ck试剂盒均购自于中生北控生物科技股份有限公司,批号分别为:20130212,20130212。
2实验方法将60只大鼠随机分成正常组、模型组、维拉帕米组(32.4mg/kg)、本发明化合物低剂量组(10mg/kg)、本发明化合物中剂量组(20mg/kg)、本发明化合物高剂量组(40mg/kg),每天按10ml/kg灌胃给药1次,连续给药7天,正常组、模型组每天给同体积纯净水,末次给药后30min,将大鼠麻醉,仰卧固定,记录标准ⅱ导联心电图。待心电图稳定后舌下静脉注射垂体后叶素0.75u/kg,10s内推注完毕,分别记录注射前(0s)及注射后第0-15min的心电图,取t波振幅升高0.1或降低0.05mv为模型成功,记录t波振幅的变化。注射垂体后叶素40min后,立即腹主动脉取血,分离血清,检测血清中sod、mda、ck、ldh水平,结果见表2。
表2大鼠急性心肌缺血中t波振幅变化
与正常组比较,△p<0.05△△p<0.01;与模型组比较,*p<0.05,**p<0.01
实验结果显示,与正常组相比,模型组t波变化值均明显高于正常组,其中在30s、1min、5min、10min四个点中与正常组比较具有统计学差异(p<0.05),表明垂体后叶素注射后能使大鼠t波振幅明显变大;与模型组相比,维拉帕米组在各点心电图中t波变化值均有明显降低,其中在30s、1min、5min三个点中与模型组比较具有统计学差异(p<0.05);本发明化合物低中高剂量均能减少t波变化幅度,其中药物低剂量组在1min时变化幅度与模型组有显著差异(分别为p<0.01,p<0.05),本发明化合物中剂量组在1min、5min、10min时变化幅度与模型组有显著差异(分别为p<0.05,p<0.01),本发明化合物高剂量组在30s、1min、5min、10min时变化幅度与模型组有显著差异(分别为p<0.01,p<0.01,p<0.05)。
表3各組大鼠血清sod、mda、ck、ldh含量水平比较
与正常组比较,△p<0.05△△p<0.01;与模型组比较,*p<0.05’**p<0.01
实验结果显示,与正常组比较,模型组mda、ck、ldh指标有统计学差异(p<0.05),sod有变化趋势但无差异(p>0.05);与模型组相比,各组sod均无统计学差异(p>0.05);维拉帕米组mda有统计学差异(p<0.05),本发明化合物低中高剂量组均能显著降低ldh水平(p<0.01),本发明化合物中高剂量组能显著升高血浆中ck水平(p<0.05),本发明化合物高剂量组能显著降低血浆mda水平(p<0.01)。
实施例6式i所示结构的化合物制备片剂药物
将350g式i所示结构的化合物和50g淀粉、7.5g羧甲基淀粉钠、0.8g滑石粉、50g糊精、0.8g硬脂酸镁及适量10%淀粉浆适混合,按照常规方法制成式i所示结构的化合物片剂1000片。每日3次,每次1片。
实施例7式i所示结构的化合物制备丸剂药物
将350g式i所示结构的化合物和12g聚乙二醇-6000、80.5g聚山梨酯-80、适量液状石蜡混合,按照常规方法制成式i所示结构的化合物丸剂1000粒。每日3次,每次1粒。
实施例8式i所示结构的化合物制备注射剂药物
将200g式i所示结构的化合物和15g注射用大豆磷脂、25g注射用甘油,注射用水定容至1000ml,按照常规方法制成式i所示结构的化合物注射剂1000支。每日1次,每次1支,至少采用250ml5%葡萄糖注射液稀释后静脉滴注。
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!