不同地区玉米粗缩病病原物的遗传变异分析方法与流程
本发明涉及分析生物学技术领域,具体涉及一种不同地区玉米粗缩病病原物的遗传变异分析方法。
背景技术:
玉米粗缩病是由水稻黑条矮缩病毒(rbsdv)引起的一种世界范围的重要病害,rbsdv能够侵染多种作物,包括水稻、玉米和小麦。各地爆发情况屡见报道,给当地玉米生产和全国的粮食安全带来非常严重的破坏。玉米粗缩病的典型症状为植株矮小,节间明显缩短,叶片显得浓绿,叶背脉上有长短不等的白色蜡泪状突起,整株叶片短脆硬直,根系不发达。
因此,明确致病原rbsdv在我国不同地区的遗传变异情况,对研究和控制各地区病害的发生意义重大。
张恒木等应用rt-pcr技术克隆了2个rbsdv中国分离物即浙江分离物和河北分离物的基因组片段s7,并测定了它们的全序列,结果表明:2个中国分离物核苷酸同源性高达99%,与日本rbsdv基因组片段s7核苷酸同源性为93.4%和93.8%,与意大利mrdvs6核苷酸同源性为85.1%和85.3%;高瑞珍等对江苏省2009年发生的水稻黑条矮缩病的病原进行了鉴定,结果表明江苏省水稻黑条矮缩病与rbsdv中国分离物相应片段的核苷酸和氨基酸序列同源性分别为93.3%~100%和97.4~100%,与srbsdv相应片段的核苷酸和氨基酸同源性为77.4~79.5%。
然而,现有技术中,致病原rbsdv在不同地区的遗传变异情况的分析仍不够准确、不够完整客观。
技术实现要素:
本发明要解决的技术问题是提供一种不同地区玉米粗缩病病原物的遗传变异分析方法,以期解决现有技术中遗传变异分析方法仍不够准确、不够完整客观的技术问题。
为解决上述技术问题,本发明采用如下技术方案:
设计一种不同地区玉米粗缩病病原物的遗传变异分析方法,包括以下步骤:
(1)取不同地区待测玉米粗缩病染病植物的叶片样本,分别提取叶片总rna;
(2)以所述总rna为模板,分别利用核苷酸序列如seqidno.2的引物进行反转录,得cdna;
(3)以所述cdna为模板,分别利用核苷酸序列如seqidno.1的引物和核苷酸序列如seqidno.2的引物进行rt-pcr;
(4)将各rt-pcr扩增产物纯化回收后测序,分析比对出可信序列;
(5)将各可信序列与rbsdv的s9序列进行比对分析样本的同源性、碱基突变、氨基酸的改变情况。
优选的,在所述步骤(3)中,rt-pcr的反应程序为:
94℃5min;94℃1min;60℃1min;72℃70s35个循环;72℃10min;4℃保存。
优选的,在所述步骤(4)中,采用clustalw2.1分析比对可信序列。
优选的,在所述步骤(5)中,采用dnaman或clustalw2.1比对分析。
与现有技术相比,本发明的有益技术效果在于:
本发明遗传变异分析方法通过扩增染病材料的rbsdvs9片段进行序列测序分析,并比对同源性,不但能够明确致病原rbsdv在我国不同地区的遗传变异情况,还可以进一步地揭示病毒的侵染传毒机制,对研究和控制各地区病害的发生意义重大。
附图说明
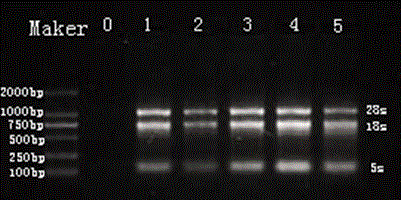
图1为不同地区发病材料中的叶片总rna电泳图谱;
图中,0孔道为ddh2o空白对照;1孔道为原阳水稻样品;2孔道为原阳玉米样品;3孔道为连云港水稻样品;4孔道为保定玉米样品;5孔道为保定小麦样品;
图2为不同地区rbsdvrt-pcr扩增后目的产物的电泳图谱;
图中,编号1为原阳水稻样品;编号2为连云港水稻样品;编号3为原阳玉米样品;编号4为保定小麦样品;编号5为保定玉米样品;
图3为五个rbsdv病毒样本的碱基和氨基酸变异分析图之一;
图4为五个rbsdv病毒样本的碱基和氨基酸变异分析图之二(续图3);
图3和4中,方框内为5处氨基酸突变。
具体实施方式
下面结合附图和实施例来说明本发明的具体实施方式,但以下实施例只是用来详细说明本发明,并不以任何方式限制本发明的范围。
在以下实施例中所涉及的仪器设备如无特别说明,均为常规仪器设备;所涉及的试剂如无特别说明,均为市售常规试剂;所涉及的试验方法,如无特别说明,均为常规方法。
实施例1:玉米粗缩病rbsdvs9序列的扩增
(1)试验材料
采集自连云港农业科学院水稻试验田(连云港)的水稻黑条矮缩病发病植株;采集河南农科院粮食作物研究所水稻试验田(原阳)的发病水稻和玉米试验田的粗缩病发病植株;采集河北农林科学院植物保护研究所实验室(保定)的绿矮病发病小麦和玉米发病幼苗。
(2)试验方法
1)总rna的提取:
剪去各样品能够明显观察到发病症状的叶片,稍多于50mg,用于叶片总rna的提取;总rna提取采用zymoresearch(usa)direct-zol™rnaminiprep试剂盒提取;
2)rt-pcr反应:
反转录所用试剂均为takara公司产品。反转录一链合成体系为10μl,如表1所示:
表1合成体系
加完样后,在pcr仪上进行下列反应:65℃5min,完成后4℃,短暂离心后,加入下列试剂完成cdna第一链的合成。
表2反应体系
加完样后放入pcr仪中反应,设定反应程序为:50℃:30min,70℃:15min,95℃:5min,4℃:hold。
反应结束后的产物即可进行pcr扩增反应。按下列样品分别加组分配制成40μlpcr反应体系,放入pcr仪中进行扩增反应。
pcr扩增用引物组合为r1-f+r1-r。结合ncbi中登记的rbsdv的s9序列(id:kc134297.1)设计引物如下:
前引物r1-f:5'-grtagacaggcaaaymtaagcgt-3';
后引物r1-r:5'-ggattacaacahacacamcgaaa-3';
表3pcr反应体系
设置pcr反应程序为:
94℃5min;94℃1min;60℃1min;72℃70s35个循环;72℃10min;4℃保存。
pcr反应结束后采用1%琼脂糖凝胶电泳检测扩增产物。因为要对目的产物进行回收测序,故琼脂糖凝胶应较厚,能够容纳每孔20μl体积的pcr反应产物。电泳条件为电压150v,20min,1×tae缓冲液,每管pcr产物平均点到两个凝胶孔中。电泳结束后开始对目的产物进行切胶回收。
(3)试验结果
对所取不同地区的五个发病材料进行叶片总rna提取,电泳上样检测后,电泳条带清晰分为三条带,分别为rna28s,18s,5s,表明rna完好无降解(如图1所示),无基因组dna污染。
分别以五种材料的rna为模板,利用设计的特异性引物组合r1-f+r1-r进行rt-pcr扩增,经琼脂糖凝胶电泳检测,目的产物约为1119bp(如图2所示),和预期结果相符。
实施例2:不同地区rbsdv病毒s9序列差异性分析
(1)将实施例1中rt-pcr扩增产物经过胶回收纯化测序后,通过clustalw2.1(multiplesequencealignment)分析软件对所测序列进行分析,共比对可信序列1083bp。
(2)通过dnaman软件将3个地区的5个病毒样本的目的核酸序列与ncbi中登记的s9序列(id:kc134297.1)进行多序列比对。
如图3和图4所示:
5个样本的同源性为97.92%,共有27处碱基发生突变,其中有5处的碱基突变后造成了氨基酸的改变,分别为80bp处的异亮氨酸突变为亮氨酸,95bp处的组氨酸突变为酪氨酸,98bp处的苯丙氨酸突变为亮氨酸,110bp处的精氨酸突变为赖氨酸,280bp处的丙氨酸突变为缬氨酸,其中80bp处、98bp处和280bp的氨基酸突变均为非极性疏水氨基酸,110bp处的氨基酸突变为碱性氨基酸突变,即突变后氨基酸的理化性质没有改变,而于95bp处的突变为碱性氨基酸变为中性氨基酸,理化性质发生了较大的改变。
此外,原阳水稻和玉米中rbsdv病毒样本与连云港水稻中rbsdv病毒样本核酸序列同源性高于河北保定小麦和玉米的rbsdv病毒样本。
综上,这些病原物属于同一种病毒在各地的变种。
上面结合附图和实施例对本发明作了详细的说明,但是,所属技术领域的技术人员能够理解,在不脱离本发明宗旨的前提下,还可以对上述实施例中的各个具体参数进行变更,形成多个具体的实施例,均为本发明的常见变化范围,在此不再一一详述。
sequencelisting
<110>河南省农业科学院
<120>不同地区玉米粗缩病病原物的遗传变异分析方法
<130>2019
<160>2
<170>patentinversion3.2
<210>1
<211>23
<212>dna
<213>人工合成
<400>1
grtagacaggcaaaymtaagcgt23
<210>2
<211>23
<212>dna
<213>人工合成
<400>2
ggattacaacahacacamcgaaa23
- 还没有人留言评论。精彩留言会获得点赞!