一种制备盐酸万古霉素杂质ImpC的方法与流程
本发明涉及一种制备盐酸万古霉素杂质impc的方法。
背景技术:
盐酸万古霉素属糖肽类抗生素,其作用机制是通过干扰细菌细胞壁结构中的关键组分肽聚糖来干扰细胞壁的合成,抑制细胞壁中磷脂和多肽的生成。
临床上用于耐甲氧西林苯青霉素的金葡菌所致的感染,以及梭状芽孢杆菌所导致的感染,及青霉素过敏者不能应用青霉素或者头孢菌素类,或者应用青霉素或者头孢菌素类无效的严重葡萄球菌感染的患者。同时也可以用于感染性心内膜炎及血液透析患者葡萄球菌感染。葡萄球菌,尤其是对耐甲氧西林金葡菌引起的败血症及重症肺部感染,盐酸万古霉素作为首选。
作为药用活性物质,有关物质对盐酸万古霉素的质量研究具有重要意义。高纯度的杂质样品对其药理毒理研究有重要意义。现有技术中已有较多关于盐酸万古霉素杂质分离方法的报道。
cn106568620b公开一种盐酸万古霉素杂质11、13和15高纯度样品的制备方法。该专利是取盐酸万古霉素结晶粉配制水溶液,然后调节ph4-6,而后水浴加热,然后回调ph至6-7,随后进行树脂层析分离,分离液体依次进行超滤和纳滤,而后进行高压液相分离,得到杂质11、13和15高纯度样品。
cn106565818a公开了一种盐酸万古霉素相关杂质4、杂质6和杂质9高纯度样品的制备方法,取盐酸万古霉素结晶粉末,配制成浓度为10~15g/l的水溶液;水浴保温30~33℃,加入乙二醇,搅拌120-122小时;然后降温到20-21℃,并缓慢滴加无水乙醇结晶;分离晶体,用高压液相色谱制备柱分离得到盐酸万古霉素中杂质4、杂质6和杂质9的高纯度样品。该方法工艺简单,大大降低了制备成本。
cn106565819a公开了一种制备盐酸万古霉素相关杂质1、杂质2和杂质10高纯度样品的方法,取盐酸万古霉素结晶粉末,配制成浓度为40~50g/l的水溶液;水浴保温25~30℃,加入双氧水,搅拌40~42小时;然后缓慢加入草酸水溶液,搅拌均匀后不再产生气泡即停止加入,然后依次进行超滤、纳滤;向纳滤液中缓慢加入乙醇进行结晶,抽滤得到结晶母液;结晶母液用高压液相色谱制备柱分离得到盐酸万古霉素中杂质1、杂质2和杂质10的高纯度样品。
cn106565820a公开了一种制备盐酸万古霉素杂质3、杂质8高纯度样品的方法。取盐酸万古霉素结晶粉末,用氯化钠水溶液配制成浓度为40-50g/l的溶液;70~75℃水浴保温72~73小时;然后降温到4~6℃,进行纳滤脱盐;纳滤液进行树脂层析富集,分别收集纯度大于80%的杂质3和杂质8的解析液;解析液依次进行超滤和纳滤;纳滤液用高压液相色谱制备柱脱盐,得到纯度大于97%的杂质3和杂质8的高纯度样品。
有关杂质impc在盐酸万古霉素成品中含量低。impc化学名为:万古霉素b糖苷配基(aglucovancomycinb),iupac名称为(3s,6r,7r,22r,23s,26s,30asa,36r,38ar)-3-(2-amino-2-oxoethyl)-10,19-dichloro-7,22,28,30,32,44-hexahydroxy-6-[[(2r)-4-methyl-2-(methylamino)pentanoyl]amino]-2,5,24,38,39-pentaoxo-2,3,4,5,6,7,23,24,25,26,36,37,38,38a-tetradecahydro-22h-8,11:18,21-dietheno-23,36-(iminomethano)-13,16:31,35-dimetheno-1h,13h-[1,6,9]oxadiazacyclohexadecino-[4,5-m][10,2,16]benzoxadiazacyclotetracosine-26-carboxylicacid。其结构式如下:
技术实现要素:
本发明的目的在于提供一种制备盐酸万古霉素杂质impc的方法。
本发明所采取的技术方案是:
一种盐酸万古霉素杂质impc的制备方法,其步骤包括:
1)取万古霉素,加入盐酸,混合后得到溶液a;
2)在溶液a中加入强酸溶液、二氯甲烷,混匀后反应16~25h,减压浓缩得到样品b;
3)溶解样品b,得到溶液c;
4)将样品c的ph值调节至7~8,反应后过滤,收集滤液,干燥,得impc粗品;
5)impc粗品经高压液相色谱制备柱分离,得到impc成品。
进一步地,所述强酸是指三氟乙酸、三氯乙酸、盐酸、磷酸中的任意一种。
优选地,所述强酸是指三氟乙酸。
进一步地,万古霉素、三氟乙酸、二氯甲烷三者质量体积比为1:(25~30):(2~3)。
进一步地,步骤2)中混匀后于50~60℃反应16~25h。
进一步地,步骤3)中,溶解样品b的溶剂是指乙酸乙酯与甲醇混合溶剂、二甲基亚砜、盐酸、氨水、甲酸中的任意一种,其中盐酸浓度为6mol/l,氨水ph9.0,甲酸ph3.0。
优选地,所述溶剂为乙酸乙酯与甲醇混合溶剂。
更优选地,乙酸乙酯与甲醇的混合体积比4:1。
进一步地,样品b与溶剂的体积比为1:(45~55)。
进一步地,步骤4)中采用naoh溶液、碳酸氢钠溶液或三乙胺溶液中的任意一种调节ph;优选地,采用naoh溶液调节ph。
进一步地,步骤5)中,高压液相色谱制备柱分离条件为:
制备分离色谱柱为:c18色谱柱,优选地,所述色谱柱为yg10ab08-020(c18)或welch
流动相a为乙腈;
流动相b为10mm的碳酸氢铵水溶液;
流速:20ml/min;
检测波长:280nm;
梯度设置如下所示:
进一步地,收集时间段45.8~47.48min的样品浓缩后脱盐,再次浓缩得impc浓缩液,将该浓缩液冻干即可得到impc成品。
通过比对目标杂质峰与主峰的相对保留时间(rrt)发现impc与已有专利中报道的化合物为不同的物质。
已有专利报道化合物rrt如下:
cn106568620b中杂质11:rrt=2.15,杂质13:rrt=2.29,杂质15:rrt=2.67;
cn106565819a中杂质1:rrt=0.43,杂质2:rrt=0.52,杂质10:rrt=1.96;
cn106565818a中杂质4:rrt=0.67,杂质6:rrt=0.8,杂质9:rrt=1.77。
impc:rrt=1.83。
本发明的有益效果是:
本发明中制备方法可以快速制备得到盐酸万古霉素相关杂质impc的高纯度样品,工艺简单,节约成本。这不仅为盐酸万古霉素的质量研究及其药理毒理研究有重要意义,同时也为进一步得到高纯度的impc奠定了基础,具有非常重要的现实意义。
附图说明
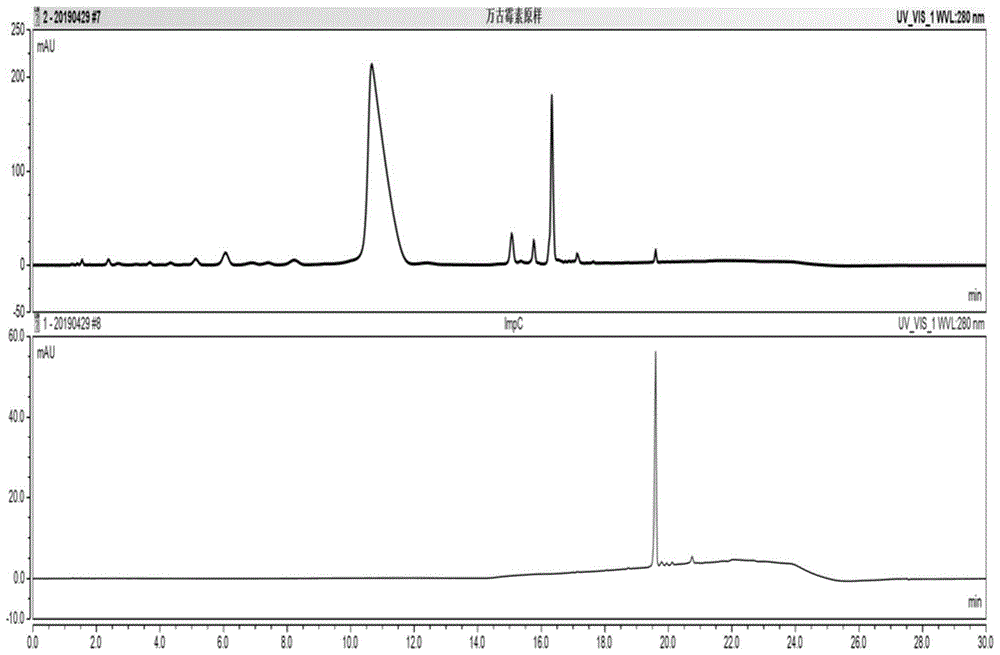
图1盐酸万古霉素原样及实施例1制备的impc成品的hplc对比分析谱图;
图2实施例1制备的impc粗品hplc液相色谱图;
图3实施例1制备的impc核磁共振氢谱图;
图4实施例1制备的impc核磁共振氢谱图;
图5实施例1制备的impc核磁共振二维h-hcosy谱图;
图6实施例2制备的impc粗品的hplc液相色谱图;
图7实施例3制备的impc粗品的hplc液相色谱图;
图8对比例1制备的impc粗品的hplc液相色谱图;
图9对比例2制备的impc粗品的hplc液相色谱图;
图10对比例3制备的impc粗品的hplc液相色谱图。
具体实施方式
下面进一步列举实施例以详细说明本发明。同样应理解,以下实施例只用于对本发明进行进一步说明,不能理解为对本发明保护范围的限制,本领域技术人员根据本发明阐述的原理做出的一些非本质的改进和调整均属于本发明的保护范围。下述示例具体的工艺参数等也仅是合适范围中的一个示例,即本领域技术人员可以通过本文的说明做合适范围内的选择,而并非要限定于下文示例的具体数据。
在以下实施例中,盐酸万古霉素由丽珠集团福州福兴制药股份有限公司提供。
在以下实施例中,盐酸万古霉素的检测方法为欧洲药典中提到的方法,采用的高压液相色谱条件如下:
高效液相色谱仪:thermou3000;
色谱柱:welchuitimatexb-c18,4.6×250mm,5μm;
流动相a:三乙胺缓冲液:乙腈:四氢呋喃=93.5:5.5:1,其中三乙胺缓冲液为0.2%三乙胺水溶液,用磷酸调节ph值至3.2;
流动相b:三乙胺缓冲液:乙腈:四氢呋喃=70:29:1,其中三乙胺缓冲液为0.2%三乙胺水溶液,用磷酸调节ph值至3.2;
梯度设置:
检测波长:λ=280nm,流速:2.0ml/min,进样体积:10μl。
实施例1
(1)取万古霉素固体粉末3g,向样品中加入6mol/l盐酸共1ml(按万古霉素重计算:每880mg约加入6mol/l盐酸0.25ml);
(2)向步骤1)得到的样品中,分别加入三氟乙酸90ml、二氯甲烷7.5ml(按万古霉素湿计算,万古霉素、三氟乙酸、二氯甲烷,三者质量体积比为1:30:2.5),后搅拌超声溶解后50~60℃放置20h;
(3)在步骤2)保温20h后,减压浓缩除去三氯乙酸,水浴温度控制30℃;
(4)向步骤3)得到的样品中,加入乙酸乙酯与甲醇混合溶剂150ml溶解,乙酸乙酯与甲醇体积比4:1;
(5)向步骤4)得到的样品溶液中,加入1mol/lnaoh溶液,调节ph至7,后将样品溶液置于4℃冰箱放置15h,取出后用砂芯漏斗减压抽滤,滤液减压干燥蒸干(水浴温度控制30℃),得所需富集样品,高压液相色谱检测富集样品,impc含量(相对百分含量)为49.61%;
(6)富集样品经高压液相色谱制备柱分离,即可得到高纯度impc样品;
制备分离条件为:
制备分离色谱柱为yg10ab08-020(c18);流动相a为乙腈;流动相b为10mm的碳酸氢铵水溶液;梯度设置如下所示:
(7)步骤6)收集时间段45.8~47.48min的样品浓缩后脱盐,后经旋转蒸发仪(水浴温度40℃)浓缩,即得目标杂质制备浓缩液,将该浓缩液经冻干机冻干即可得到impc成品。
经检测,impc纯度可达97%。
将实施例1制备的impc成品溶液、原样溶液进行hplc对比分析,确认两者的hplc保留特征均一致(图1)。
图2impc粗品hplc液相色谱图。
图3为实施例1中制备的impc核磁共振氢谱图,由该图可知impc对应的氢谱信息。
图4为实施例1中制备的impc核磁共振碳谱图,由该图可知impc对应的碳谱信息。
图5为实施例1中制备的impc核磁共振二维h-hcosy谱图,由该图可知impc氢谱中是否有2键或3键质子自旋-自旋耦合(j-coupling)存在。
实施例2
(1)取万古霉素固体粉末3g,向样品中加入6mol/l盐酸共1ml(按万古霉素重计算:每880mg约加入6mol/l盐酸0.25ml);
(2)向步骤1)得到的样品中,分别加入三氟乙酸90ml、二氯甲烷7.5ml(按万古霉素湿计算,万古霉素、三氟乙酸、二氯甲烷,三者质量体积比为1:30:2.5),后搅拌超声溶解后50~60℃放置16h;
(3)在步骤2)保温20h后,减压浓缩除去三氯乙酸,水浴温度控制30℃;
(4)向步骤3)得到的样品中,加入乙酸乙酯与甲醇混合溶剂150ml溶解,乙酸乙酯与甲醇体积比4:1;
(5)向步骤4)得到的样品溶液中,加入1mol/lnaoh溶液,调节ph至7,后将样品溶液置于4℃冰箱放置15h,取出后用砂芯漏斗减压抽滤,滤液减压干燥蒸干(水浴温度控制30℃),得所需富集样品,高压液相色谱检测富集样品,impc含量(相对百分含量)为49.61%;
(6)富集样品经高压液相色谱制备柱分离,即可得到高纯度impc样品;
制备分离条件为:
制备分离色谱柱为yg10ab08-020(c18);流动相a为乙腈;流动相b为10mm的碳酸氢铵水溶液;梯度设置如下所示:
(7)步骤6)收集时间段45.8~47.48min的样品浓缩后脱盐,后经旋转蒸发仪(水浴温度40℃)浓缩,即得目标杂质制备浓缩液,将该浓缩液经冻干机冻干即可得到impc成品。
经检测,impc纯度可达96.7%。
图6为impc粗品制备液相色谱图,由图6可知:实施例2酸破坏时间合适,已达到破坏目的,且二者目标组分含量及相关杂质无明显差异。
实施例3
(1)取万古霉素固体粉末3g,向样品中加入6mol/l盐酸共1ml(按万古霉素重计算:每880mg约加入6mol/l盐酸0.25ml);
(2)向步骤1)得到的样品中,分别加入三氟乙酸90ml、二氯甲烷7.5ml(按万古霉素湿计算,万古霉素、三氟乙酸、二氯甲烷,三者质量体积比为1:30:2.5),后搅拌超声溶解后50~60℃放置25h;
(3)在步骤2)保温20h后,减压浓缩除去三氯乙酸,水浴温度控制30℃;
(4)向步骤3)得到的样品中,加入乙酸乙酯与甲醇混合溶剂150ml溶解,乙酸乙酯与甲醇体积比4:1;
(5)向步骤4)得到的样品溶液中,加入1mol/lnaoh溶液,调节ph至7,后将样品溶液置于4℃冰箱放置15h,取出后用砂芯漏斗减压抽滤,滤液减压干燥蒸干(水浴温度控制30℃),得所需富集样品,高压液相色谱检测富集样品,impc含量(相对百分含量)为49.61%;
(6)富集样品经高压液相色谱制备柱分离,即可得到高纯度impc样品;
制备分离条件为:
制备分离色谱柱为yg10ab08-020(c18);流动相a为乙腈;流动相b为10mm的碳酸氢铵水溶液;梯度设置如下所示:
(7)步骤6)收集时间段45.8~47.48min的样品浓缩后脱盐,后经旋转蒸发仪(水浴温度40℃)浓缩,即得目标杂质制备浓缩液,将该浓缩液经冻干机冻干即可得到impc成品。
经检测,impc纯度可达97.1%。
图7为impc粗品制备液相色谱图,由图7可知:实施例3酸破坏时间合适,已达到破坏目的,且二者目标组分含量及相关杂质无明显差异。
对比例1
对比例1的分离纯化步骤同实施例1,不同之处在于步骤2)中的保温时间为:4h,也就是说步骤2)的具体步骤如下:
向步骤1)得到的样品中,分别加入三氟乙酸90ml、二氯甲烷7.5ml(按万古霉素重计算,万古霉素、三氟乙酸、二氯甲烷,三者质量体积比为1:30:2.5),后搅拌超声溶解后50~60℃分别放置4h。
图8为impc粗品制备液相色谱图。
对比例2
对比例2的分离纯化步骤同实施例1,不同之处在于步骤2)中的保温时间为:10h,也就是说步骤2)的具体步骤如下:
向步骤1)得到的样品中,分别加入三氟乙酸90ml、二氯甲烷7.5ml(按万古霉素重计算,万古霉素、三氟乙酸、二氯甲烷,三者质量体积比为1:30:2.5),后搅拌超声溶解后50~60℃分别放置10h。
图9为impc粗品制备液相色谱图。
对比例3
对比例5的分离纯化步骤同实施例1,不同之处在于步骤2)中的保温时间为:30h,也就是说步骤2)的具体步骤如下:
向步骤1)得到的样品中,分别加入三氟乙酸90ml、二氯甲烷7.5ml(按万古霉素重计算,万古霉素、三氟乙酸、二氯甲烷,三者质量体积比为1:30:2.5),后搅拌超声溶解后50~60℃分别放置30h。
图10为impc粗品制备液相色谱图。
由图8和图9可知:对比例1、2酸破坏时间较短,没有破坏完全,其中由对比例1液相色谱图8可明显看出盐酸万古霉素含量较高(主成分),且同时有另一未知主要杂质存在,目标杂质含量极低;由对比例2液相色谱图9可明显看出盐酸万古霉素已不可见,目标杂质含量相对对比例1来说,含量在增高,但同时有另一未知主要杂质存在。由此可说明经对比例1、2处理后,目标杂质含量仍然很低,酸破坏时间过短,未达到完全破坏的目的。
由图10可知,对比例3中酸破坏时间较长,目标成分已极低及相关杂质数目较多,已失去酸破坏意义。
- 还没有人留言评论。精彩留言会获得点赞!