一种以2-氨基-5-甲基苯甲酸为原料高效合成Cetilistat的工艺的制作方法
本发明涉及一种已知化合物的合成方法,具体地说是一种以2-氨基-5-甲基苯甲酸为原料高效合成cetilistat的工艺,属于有机合成技术领域。
背景技术:
肥胖是现代社会常见的营养性问题,是体内脂肪,尤其是甘油三脂积聚过多而导致的一种状态,易引发心脏病、高血压、糖尿病等疾病。cetilistat(赛利司他)是由norgine和武田共同开发,英文商品名为oblean,于2013年9月20日获日本医药品医疗器械综合机构(pmda)批准,由武田(takeda)在日本上市销售,用于治疗肥胖。赛利司他本质上一种长效和强效的特异性胃肠道脂肪酶抑制剂,通过与胃和小肠腔内胃脂肪酶和胰脂肪酶的活性丝氨酸部位形成共价键使酶失活而发挥治疗作用,失活的酶不能将食物中的脂肪(主要是甘油三酯)水解为可吸收的游离脂肪酸和单酰基甘油,未消化的甘油三酯不能被身体吸收,从而减少热量摄入,控制体重。该药最大优点是不作用于神经系统,不影响胃肠道的其他酶活性,与现行的同类药物奥利司他的作用机制相似,但与后者相比,排泄失禁及胃肠胀气等不良反应发生率低,具有更好的安全性和耐受性。其结构式见下式1:
cetilistat原料药的合成制备方法在有关专利和论文中均有报道,主要有以下几种方法:
方法一:在专利cn1359378a中公开了上述结构化合物及其相关的制备方法
(1)
(2)
上述制备路线以2-氨基-5-甲基苯甲酸与氯甲酸正十六烷醇酯反应,一步达到氨基酰化、羧基活化和环化反应合成赛利司他的目的。该合成路线简单,但实际合成反应中需要使用大过量的氯甲酸正十六烷醇酯才能完成这些转化,过量的试剂反应产物中含有大量十六烷醇等杂质导致后续的分离纯化困难,实际收率大大降低,总收率只有15%。并且难以有效解决正十六烷醇酯在产品中的残留问题,工业化实施受到限制。
制备路线(2)是在路线(1)基础上的一种改进,将反应分阶段进行:先将2-氨基-5-甲基苯甲酸与过量约15%的氯甲酸正十六烷醇酯反应,进行氨基的酰化,再与氯甲酸甲酯反应进行羧基的活化、环合。经过这样的改进减少了氯甲酸正十六烷醇酯的用量,总收率提高到31%。相比与路线(1),路线(2)收率有了一定的改善,但是该反应副反应多,单步转化率低,中间体不易分离纯化,最终的cetilistat收率低,杂质含量高。
方法二:在专利cn1785967a中公开了另外一条制备路线:
以对甲苯基异氰酸酯为起始原料与十六烷醇反应得到十六烷基对甲苯基氨基甲酸酯(收率57%),再与溴发生苯环溴代(收率74%)反应,在双三苯基膦二氯化钯催化下co和水与卤代芳环缩合引入羧基得到西替利司它前体氨基甲酸酯羧基物(收率77%),再在氯甲酸乙酯作用下环合得到目标产物cetilistat(收率82%),总收率约27%。该条路线长,副反应多,重复性差,总收率低,使用的金属催化剂三苯基膦二氯化钯价格昂贵,第三步反应需要高温(115℃)、高压(8bar)等条件,对设备要求较高,不利于产业化。
方法三:在专利cn103936687a中公布了一种制备方法:
以2-氨基-5-甲基苯甲酸为原料,经羧基酯化,氨基酰化,脱脂化和环化四步反应。该发明技术方案和上述提及的几种方法相比,该方法反应条件相对温和,解决了正十六烷醇酯残留的问题,总收率提高到50%(以2-氨基-5-甲基苯甲酸计)。但是该工艺路线长,副反应较多,中间体不易分离纯化,单步收率低,尤其是第一步转化率非常低。另外,此路线只适合在实验室小批量合成,对于工业化大量制备受到限制。
综上所述,目前已有的方法存在收率低、成本高、产品难提纯、不易工业化生产这些问题。因此,开发一条收率高,重复性好,绿色经济的合成路线,具有非常重大的意义。
技术实现要素:
本发明针对目前赛利司他(十六烷氧基-6-甲基-4h-3,1-苯并噁嗪-4-酮)合成技术存在问题,提供了一种以2-氨基-5-甲基苯甲酸为原料高效合成cetilistat的工艺。本发明合成cetilistat反应步骤少,工艺路线短,副反应少,最终产品收率高,纯度>99%。
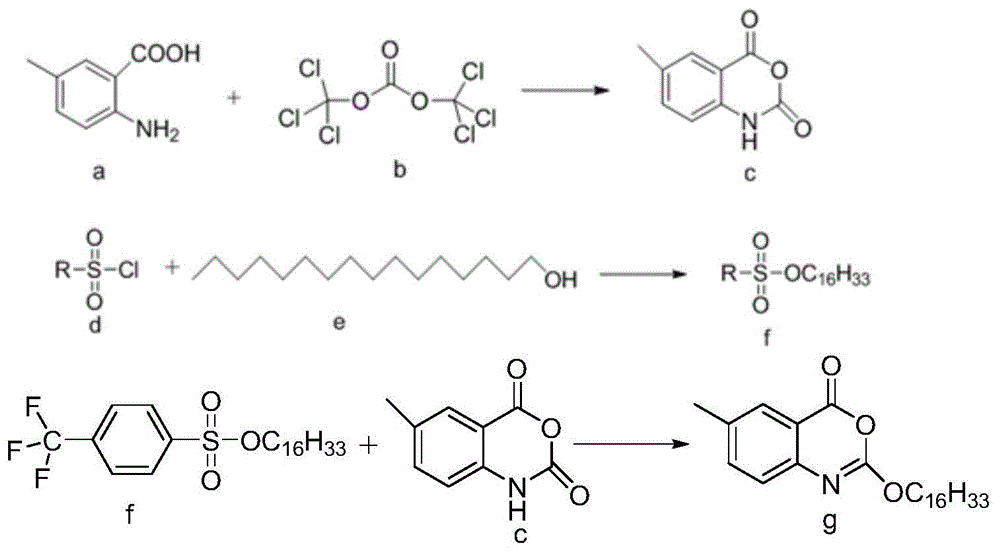
本发明以2-氨基-5-甲基苯甲酸为原料高效合成cetilistat的工艺,是以2-氨基-5-甲基苯甲酸(a)为原料,与三光气(b)在有机碱催化的作用下发生酯交换反应生成6-甲基-2,4-二氢-1h--3,1-苯并恶嗪-2,4-二酮(中间体c);再采用特定结构的磺酰氯(d),在碱催化的条件下与正十六醇(e)反应生成磺酸酯(中间体f);最后,中间体c和中间体f在有机碱存在下发生氧原子上的烷基化反应生成目标产物cetilistat。具体包括如下步骤:
步骤1:酯交换反应合成中间体c
将原料2-氨基-5-甲基苯甲酸(a)与三光气(b),在适量有机碱的存在下,于溶剂中加热缩合获得中间体c——6-甲基-2,4-二氢-1h--3,1-苯并恶嗪-2,4-二酮;
步骤1中,所述有机碱选自吡啶、三乙胺、二异丙基乙基胺、n,n-二甲基苯胺、tmg、dbu或dbn等路易斯碱进行催化。
步骤1中,所述溶剂优选使用非质子性溶剂,如四氢呋喃、乙腈、二氯甲烷、乙酸乙酯、dmf、乙酸丁酯、氯仿、丙酮、丁酮等,既可以使用单一溶剂也可以使用混合溶剂。
步骤1中,原料2-氨基-5-甲基苯甲酸(a)与三光气(b)的摩尔比为1:1-6:1,优选3:1;原料a与有机碱的摩尔比为1:1-5.5,优选1:2。
步骤1中,本步骤的反应温度为20-60℃,优选52℃。
本步骤的反应过程如下:
步骤2:中间体f磺酸酯的制备
将过量的磺酰氯和正十六烷醇依次溶解在溶剂中,加入碱作为缚酸剂,加热回流反应;反应完成后分别用稀盐酸、氢氧化钠溶液、水等将有机相洗涤至中性,再用二氯甲烷萃取,干燥,减压蒸去溶剂,得纯品f。
所述磺酰氯的结构式为:
其中r为烃基或芳香基,其中烃基为十个碳原子以下的饱和烃基或不饱和烃基,饱和烃基如甲基、乙基、正丙基、正丁基、正戊基、异丙基、叔丁基、异戊基、新戊基等;不饱和烃基如甲烯基、乙烯基、乙炔基、烯丙基、炔丙基、己烯基等;芳香基如:苯基、甲基苯基、乙基苯基、丙基苯基、硝基苯基、卤代苯基、三氟甲基苯基等;本发明优选苯磺酰氯。
所述碱可以为有机碱或无机碱,所述有机碱为叔胺或季铵碱,选自三甲胺、三乙胺、三丙胺、二异丙基乙基胺、n,n-二甲基苯胺、吡啶、4-二甲氨基吡啶等,所述无机碱选自碱金属以及碱土金属的氧化物、氢氧化物、氢化物、碳酸盐、碳酸氢盐、羧酸盐等,既可以用单一碱催化,也可以用两种混合碱催化,如4-二甲氨基吡啶和三乙胺体系,三乙胺-二异丙基乙基胺体系等。
所述溶剂优先使用非质子溶剂,如四氢呋喃、乙腈、二氯甲烷、乙酸乙酯、乙酸甲酯、dmf、nmp、dmac、乙酸丁酯、氯仿、丙酮、丁酮、甲苯、二甲苯、氯苯、二氯苯、苯甲醚、二苯醚等,既可以使用单一溶剂也可以使用混合溶剂。
步骤2中,磺酰氯与正十六烷醇的摩尔比为1:8-1,优选1.5:1;十六烷醇与碱的摩尔比为1:2-10,优选1:2.2。
步骤2中,反应温度为20-100℃,优选40-80℃。
本步骤的反应过程如下:
步骤3:目标产物的制备
中间体f和中间体c在适量碱的催化下,于溶剂中加热回流反应,反应结束后减压蒸去溶剂,所得粗品再用乙醇:乙酸乙酯=9:1重结晶即得cetilistat。
本步骤中,选择碱性稍强的有机碱催化,如三乙胺、三甲胺、甲基吡啶、4-二甲氨基吡啶、n-甲基哌啶、咪唑、n-甲基吗啉、氢氧化四甲基铵、吡啶、三乙胺等。
本步骤中,低沸点的非质子性溶剂对反应有利,因此所述溶剂选自二氯甲烷、乙酸乙酯、乙醚、乙腈或四氢呋喃等,优选二氯甲烷。
本步骤中,反应温度为20-50℃,优选40℃;反应时间为10-12h,优选10.5h。
本步骤中,中间体c与有机碱的摩尔比为1:1-4,优选1:1.8;中间体c和中间体f的摩尔比为1:1-2.5,优选1:1.6。
本步骤的反应过程如下:
本发明的有益效果体现在:
1、反应路线短,收率高。该合成路线共三步,每一步收率都在82%以上,合计总收率不低于52%远高于目前已有的方法。
2、反应条件温和,后处理简单,重复性好,有一定工业生产价值。本发明提出了一种全新的cetilistat的合成方法,每一步的反应温度都在100℃以内,无需加压,常压下即可以发生反应;反应后容易处理,利用常用的柱层析和重结晶的方法,重复性好,有一定的工业生产价值。
3、不使用金属类催化剂,产品中不会有金属残留的问题,产品的质量有保证。
附图说明
图1是本发明合成路线示意图。
具体实施方式
实施例1:
1、中间体c(6-甲基-2,4-二氢-1h--3,1-苯并恶嗪-2,4-二酮)的合成
在250ml反应瓶中投入2-氨基-5-甲基苯甲酸(4g,0.026mol),加入150ml乙腈,油浴锅加热到52℃,三光气(2.57g,0.0087mol)先用40ml的乙腈溶解,同时向反应瓶内缓慢滴加三光气和吡啶(4.1g,0.052mol),在52℃下搅拌5h,减压除去溶剂后,加入h2o(100ml),抽滤,滤饼依次用水和冰二氯甲烷洗涤,真空干燥,得产品4.27g,收率93%。1hnmr(400mhz,d6-dmso):δ11.63(s,1h),7.72(s,1h),7.58.7.55(dd,j1=4.0hz,j2=8.0hz,1h),7.06(d,j=8.0hz,1h),2.33(s,3h)
2、中间体f(磺酸酯)的制备
在250ml反应瓶中加入十六烷醇(5g,0.021mol),150ml二氯甲烷溶解,再依次加入对三氟甲基苯磺酰氯(6.6g,0.022mol)、吡啶(3.8g,0.0484mol),dmap(0.18g,1.5mmol),升温回流12h,反应完成后,依次用50ml的1mol/l的盐酸,50ml的1%的naoh,50ml的水洗涤反应液至中性,分液,水层依次用40ml的二氯甲烷萃取三次,合并有机相,无水硫酸钠干燥,抽滤,真空除去溶剂,得米白色固体8.01g,收率97%。1hnmr(400mhz,cdcl3):δ7.81(d,j=8.4hz,2h),7.73(d,j=8.0hz,2h),4.02(m,j=6.4hz,2h),1.69-1.64(m,3h),1.33-1.15(m,25h),0.84(t,j=7.0hz,3h)
3、cetilistat的制备
冰浴条件下在250ml反应瓶中加入中间体6-甲基-2,4-二氢-1h--3,1-苯并恶嗪-2,4-二酮(4g,0.023mol),加入200ml无水二氯甲烷溶解,然后依次滴加对三氟甲基苯磺酸十六烷基酯(12.4g,0.0276mol)的二氯甲烷溶液、三乙胺(4.18g,0.0414mol),滴加完毕后,升温回流,反应完毕后,减压蒸除溶剂,所得粗品再用乙醇:乙酸乙酯=9:1重结晶得纯品cetilistat10g收率90%。1hnmr(400mhz,cdcl3):δ7.93(s,1h),7.55(dd,j=8.4,8.0hz,1h),7.34(d,j=8.4hz,1h),4.44(t,j=6.4hz,2h),2.44(s,3h),1.85-1.78(m,2h),1.48~1.45(m,2h),1.401.28(m,21h),0.90(t,j=6.4hz,3h)
实施例2:
1、中间体c(6-甲基-2,4-二氢-1h--3,1-苯并恶嗪-2,4-二酮)的合成
在250ml反应瓶中投入2-氨基-5-甲基苯甲酸(4g,0.026mol),加入150ml乙腈,三光气(2.57g,0.0087mol)先用40ml的乙腈溶解,同时向反应瓶内缓慢滴加三光气(2.57g,0.0087mol)和吡啶(4.1g,0.052mol),室温条件下5h,减压除去溶剂后,加入h2o(100ml),抽滤,滤饼依次用水和冰二氯甲烷洗涤,真空干燥,得产品3.0g,收率65%。1hnmr(400mhz,d6-dmso):δ11.63(s,1h),7.72(s,1h),7.58.7.55(dd,j1=4.0hz,j2=8.0hz,1h),7.06(d,j=8.0hz,1h),2.33(s,3h),1.69-1.64(m,3h),1.33-1.15(m,25h),0.84(t,j=7.0hz,3h)
中间体f及终产物g的合成同实施例1。
实施例3:
1、中间体c的合成同实施例1;
2、中间体f(磺酸酯)的制备
在250ml反应瓶中加入对十六烷醇(5g,0.021mol),加入二氯甲烷150ml溶解,再依次加入对甲基苯磺酰氯(5.5g,0.028mol)、吡啶(4.8g,0.0616mol),dmap(0.18g,1.5mmol),滴加完毕后,升温回流12h,反应完成后,依次用50ml的1mol/l的盐酸,50ml的1%的naoh,50ml的水洗涤反应液至中性,分液,水层依次用40ml的二氯甲烷萃取三次,合并有机相,无水硫酸钠干燥,抽滤,真空除去溶剂,得米白色固体5.95g,收率72%。1hnmr(400mhz,cdcl3):d7.78(d,j=8.4hz,2h),7.34(d,j=8.0hz,2h),4.02(t,j=6.4hz,2h),2.44(s,3h),1.69-1.64(m,3h),1.33-1.15(m,25h),0.84(t,j=7.0hz,3h)
3、cetilistat的制备
冰浴条件下在150ml反应瓶中加入中间体c(2g,0.011mol),加入80ml无水二氯甲烷溶解,然后依次滴加中间体(3.63g,0.0276mol)的二氯甲烷溶液,三乙胺(2.0g,0.0198mol)滴加完毕后,升温回流,反应完毕后,减压蒸除溶剂,所得粗品再用乙醇:乙酸乙酯=9:1重结晶得纯品cetilistat2.2g,收率50%。
1hnmr(400mhz,cdcl3):δ7.93(s,1h),7.55(dd,j=8.4,8.0hz,1h),7.34(d,j=8.4hz,1h),4.44(t,j=6.4hz,2h),2.44(s,3h),1.85-1.78(m,2h),1.48~1.45(m,2h),1.401.28(m,21h),0.90(t,j=6.4hz,3h)
实施例4:
1、中间体c的合成同实施例1;
2、中间体f(磺酸酯)的制备:
在250ml反应瓶中加入对十六烷醇(5g,0.021mol),加入二氯甲烷150ml溶解,再依次加入对甲基苯磺酰氯(5.5g,0.028mol)、naoh(2.5g,0.0616mol),滴加完毕后,升温回流12h,反应结束后,依次用50ml的1mol/l的盐酸,50ml的1%的naoh,50ml的水洗涤反应液至中性,分液,水层依次用40ml的二氯甲烷萃取三次,合并有机相,无水硫酸钠干燥,抽滤,真空除去溶剂,得米白色固体4.9g,收率60%。1hnmr(400mhz,cdcl3):d7.78(d,j=8.4hz,2h),7.34(d,j=8.0hz,2h),4.02(t,j=6.4hz,2h),2.44(s,3h),1.69-1.64(m,3h),1.33-1.15(m,25h),0.84(t,j=7.0hz,3h)
3、终产物g的合成同实施例2。
- 还没有人留言评论。精彩留言会获得点赞!