一种吲哚衍生物的制备方法与流程
本发明属于药物技术领域,尤其涉及一种吲哚衍生物的制备方法。
背景技术:
赛洛多辛中间体杂质5-[(2r)-2-氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚盐是吲哚衍生物的一种。赛洛多辛化学名称为1-(3-羟丙基)-5-[(2r)-2-({2-(2,2,2-三氟乙氧基)苯氧基]乙基}氨基)丙基]-2,3-二氢-1h-吲哚-7-羧基酰胺,是由日本原研橘生和日本第一三工制药联合研发销售的新一代对α1-肾上腺受体阻滞剂,对α1a-亚型的肾上腺素受体具有突出的选择性,能够选择性松弛尿道平滑肌。它还可以降低了低血压的发生率,可用于治疗与良性前列腺增生(bph)或肥大引起的排尿困难的症状。在制备赛洛多辛时,脱氢杂质的含量比较大,并且此杂质5-[(2r)-2-氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚盐和赛洛多辛结构十分类似,且该产物是油状物,纯度低,在成品中比较难纯化,而杂质的存在,不仅会影响赛洛多辛的纯度,还可能带来毒副作用。
技术实现要素:
本发明的目的在于克服现有技术的上述不足,提供一种吲哚衍生物的制备方法,旨在提高获取的赛洛多辛中间体杂质的纯度。
为实现上述发明目的,本发明采用的技术方案如下:
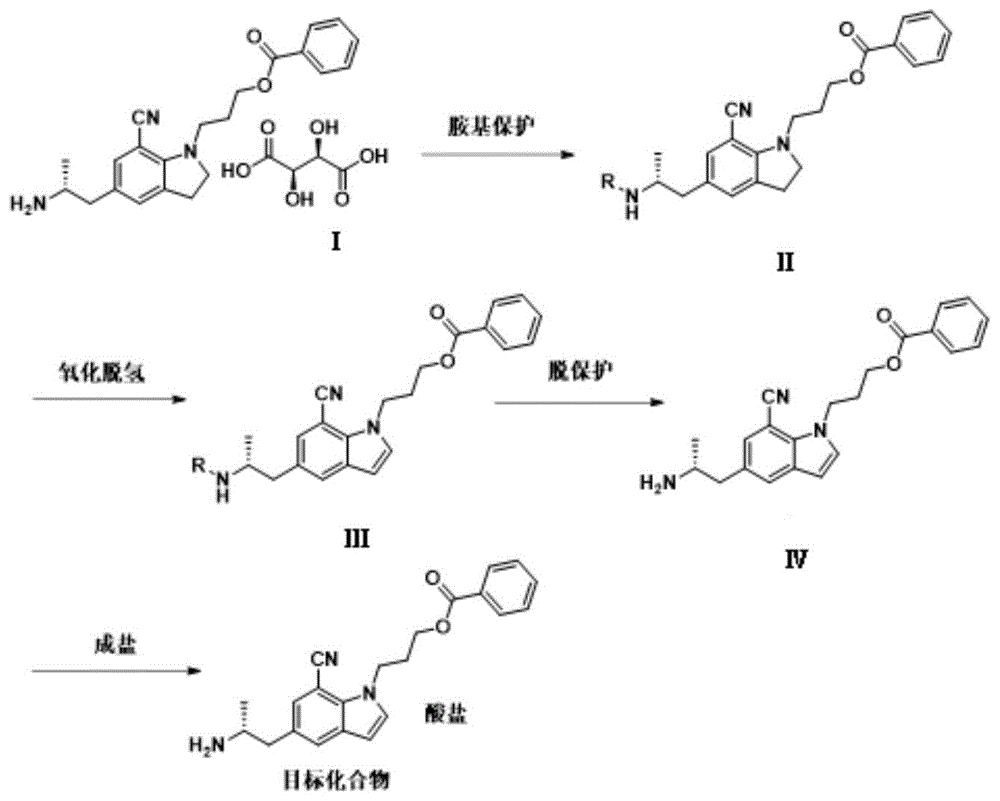
本发明提供一种吲哚衍生物的制备方法,所述制备方法包括:
将化合物ⅰ和碱加入第一反应溶剂里,搅拌溶清后再加入氨基保护剂进行反应,反应完成后,在有机相中干燥浓缩得到化合物ⅱ,所述化合物ⅰ为5-[(2r)-2-氨基丙基]-1-[3-(苯甲酰氧基)丙基]-2,3-二氢-7-氰基-1h-吲哚酒石酸盐;
将所述化合物ⅱ加入第二反应溶剂中,搅拌溶清后加入氧化剂进行反应得到化合物ⅲ;
将所述化合物ⅲ加入到第三反应溶剂中,用脱保护试剂进行氨基的脱保护反应,得到化合物iv;
将所述化合物iv和酸加入到第四反应溶剂中进行反应,得到目标化合物,所述目标化合物为5-[(2r)-2-氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚盐。
在一个实施例中,所述化合物ⅰ的结构式为:
化合物ⅱ的结构式为:
化合物ⅲ的结构式为:
化合物iv的结构式为:
目标化合物的结构式为:
在一个实施例中,所述碱包括三乙胺、二异丙胺、叔丁醇钾、氢氧化钠、碳酸钾、碳酸钠中的至少一种;
和/或,所述酸包括酒石酸、盐酸、乙二酸、硫酸、丙二酸、乙酸、柠檬酸、马来酸、富马酸中的至少一种。
在一个实施例中,所述第一反应溶剂包括四氢呋喃、二氯甲烷、乙酸乙酯、甲醇、乙醇、二甲基甲酰胺、水、异丙醇,最优选为四氢呋喃、二氯甲烷、二甲基甲酰胺中的至少一种;
和/或,所述第二反应溶剂包括四氢呋喃、二氯甲烷、乙酸乙酯、甲醇、乙醇、二甲基甲酰胺、水、异丙醇中的至少一种;
和/或,所述第三反应溶剂包括二氯甲烷,乙酸乙酯,四氢呋喃,甲醇,乙醇中的至少一种;
和/或,所述第四反应溶剂包括四氢呋喃、二氯甲烷、乙酸乙酯、甲醇、乙醇、异丙醇中的至少一种。
在一个实施例中,所述氨基保护剂包括叔丁氧羰基、苄氧羰基、2-联苯基-2-丙氧羰基、邻苯二甲酰亚胺基、对甲苯磺酰基、三苯甲基、甲酰基、三氟乙酰基、笏甲氧羰基中的至少一种。
在一个实施例中,所述氧化剂包括二氧化锰、2,3-二氯-5,6-二氰对苯醌(ddq)、硝酸、重铬酸钾、高锰酸钾、间氯过氧苯甲酸中的至少一种。
在一个实施例中,所述脱保护试剂包括钯碳/氢气、三氟乙酸、盐酸、四丁基氟化铵、三乙烷基铵甲酸盐、氢溴酸、乙酸中的至少一种。
在一个实施例中,所述化合物ⅰ与所述胺基保护剂的摩尔比为1:0.5~2.0;
和/或,所述化合物ⅱ与所述氧化剂的摩尔比为1:0.5~30。
在一个实施例中,所述将化合物ⅰ和碱加入第一反应溶剂里,搅拌溶清后再加入氨基保护剂进行反应,反应完成后,在有机相中干燥浓缩得到化合物ⅱ步骤中,搅拌反应时间为1h~72h,搅拌反应温度为0℃~70℃;
和/或,所述将所述化合物ⅱ加入第二反应溶剂中,搅拌溶清后加入氧化剂进行反应得到化合物ⅲ步骤中,反应时间为1h~48h,反应温度为0℃~120℃;
和/或,所述将所述化合物ⅲ加入到第三反应溶剂中,用脱保护试剂进行氨基的脱保护反应,得到化合物iv步骤中,反应时间为1h~48h,反应温度为0℃~120℃;
和/或,所述将所述化合物iv和酸加入到第四反应溶剂中进行反应,得到目标化合物步骤中,反应时间为0.5h~12h,反应温度为20℃~120℃。
在一个实施例中,所述将所述化合物iv和酸加入到第四反应溶剂中进行反应,得到目标化合物步骤中,将所述化合物iv和酸加入到第四反应溶剂中进行反应后,通过加热回流溶清,冷却降温析晶,再干燥得到所述目标化合物。
在一个实施例中,析晶时间为1h~48h,干燥温度可为20~100℃,干燥时间为1h~72h。
通过本发明提供的吲哚衍生物的制备方法制备的目标化合物中,赛洛多辛中间体脱氢杂质的标定含量大于95.0%,可作为杂质标准品,应用于赛洛多辛原料及其制剂的杂质定性、定量研究,对有效控制赛洛多辛原料及其制剂的质量具有重要意义。
附图说明
图1为本发明的制备路线;
图2为本发明实施例1制得的产物的高效液相色谱谱图;
图3为本发明实施例1制得的产物的质谱谱图;
图4为本发明实施例1制得的产物的核磁共振氢谱谱图;
图5为本发明实施例3制得的高效液相色谱谱图。
具体实施方式
为了使本发明要解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
本发明实施例一种吲哚衍生物的制备方法,包括如下步骤:
步骤s10:将化合物ⅰ和碱加入第一反应溶剂里,搅拌溶清后再加入氨基保护剂进行反应,反应完成后,在有机相中干燥浓缩得到化合物ⅱ,所述化合物ⅰ为5-[(2r)-2-氨基丙基]-1-[3-(苯甲酰氧基)丙基]-2,3-二氢-7-氰基-1h-吲哚酒石酸盐;
步骤s20:将所述化合物ⅱ加入第二反应溶剂中,搅拌溶清后加入氧化剂进行反应得到化合物ⅲ;
步骤s30:将所述化合物ⅲ加入到第三反应溶剂中,用脱保护试剂进行氨基的脱保护反应,得到化合物iv;
步骤s40:将所述化合物iv和酸加入到第四反应溶剂中进行反应,得到目标化合物,所述目标化合物为5-[(2r)-2-氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚盐。
本发明提供的吲哚衍生物的制备方法,赛洛多辛中间体脱氢杂质的标定含量大于95.0%,可作为杂质标准品,应用于赛洛多辛原料及其制剂的杂质定性、定量研究,对有效控制赛洛多辛原料及其制剂的质量具有重要意义。
进一步地,步骤s10中,碱包括三乙胺、二异丙胺、叔丁醇钾、氢氧化钠、碳酸钾、碳酸钠中的至少一种,优选为三乙胺。
第一反应溶剂包括四氢呋喃、二氯甲烷、乙酸乙酯、甲醇、乙醇、二甲基甲酰胺、水、异丙醇中的至少一种,优选包括四氢呋喃、二氯甲烷、二甲基甲酰胺中的至少一种。
氨基保护剂包括叔丁氧羰基、苄氧羰基、2-联苯基-2-丙氧羰基、邻苯二甲酰亚胺基、对甲苯磺酰基、三苯甲基、甲酰基、三氟乙酰基、笏甲氧羰基中的至少一种;化合物ⅰ与氨基保护剂的摩尔比为1:0.5~2.0,例如可以为1:0.5、1:1.0、1:1.5、1:2.0等,优选为1:1.0~1.3。
搅拌溶清后再加入氨基保护剂进行反应的过程中,搅拌反应时间为1h~72h,例如可以为1h、2h、3h、6h、12h、18h、24h、30h、36h、48h、60h、72h等,优选为1h~3h;搅拌反应温度可为0℃~70℃,例如可以为0℃、5℃、10℃、15℃、20℃、25℃、30℃、35℃、40℃、45℃、50℃、55℃、60℃、65℃、70℃等,优选为10~30℃。
进一步地,步骤s20中,第二反应溶剂包括四氢呋喃、二氯甲烷、乙酸乙酯、甲醇、乙醇、二甲基甲酰胺、水、异丙醇中的至少一种,优选为二氯甲烷或乙酸乙酯。
氧化剂包括二氧化锰、2,3-二氯-5,6-二氰对苯醌(ddq)、硝酸、重铬酸钾、高锰酸钾、间氯过氧苯甲酸中的至少一种,优选为2,3-二氯-5,6-二氰对苯醌(ddq)或二氧化锰。化合物ⅱ与氧化剂的摩尔比为1:0.5~30,例如可以为1:0.5、1:1.0、1:2.0、1:3.0、1:5.0、1:10、1:15、1:20、1:25、1:30等,优选为1:2~10。
将所述化合物ⅱ加入第二反应溶剂中,搅拌溶清后加入氧化剂进行反应的过程中,反应时间为1h~48h,例如可以为1h、2h、3h、6h、12h、18h、24h、30h、36h、42h、48h等,优选为12h~24h;反应温度为0℃~120℃,例如可以为0℃、5℃、10℃、15℃、20℃、25℃、30℃、35℃、40℃、50℃、60℃、80℃、100℃、120℃等,优选为10℃~40℃。
进一步地,步骤s30中,第三反应溶剂包括二氯甲烷,乙酸乙酯,四氢呋喃,甲醇,乙醇中的至少一种,优选为二氯甲烷或乙酸乙酯。
脱保护试剂可选择包括钯碳/氢气、三氟乙酸、盐酸、四丁基氟化铵、三乙烷基铵甲酸盐、氢溴酸、乙酸中的至少一种,优选为三氟乙酸或盐酸。
反应时间为1h~48h,例如可以为1h、2h、3h、6h、12h、18h、24h、30h、36h、42h、48h等,优选为4~6h;反应温度为0℃~100℃,例如可以为0℃、5℃、10℃、15℃、20℃、25℃、30℃、35℃、40℃、50℃、60℃、80℃、100℃等,优选为10℃~30℃。
进一步地,步骤s40中,第四反应溶剂包括四氢呋喃、二氯甲烷、乙酸乙酯、甲醇、乙醇、异丙醇中的至少一种,优选为乙酸乙酯或异丙醇。
酸包括酒石酸、盐酸、乙二酸、硫酸、丙二酸、乙酸、柠檬酸、马来酸、富马酸中的至少一种,优选为乙二酸或酒石酸。
反应时间为0.5h~12h,例如可以为0.5h、1h、2h、4h、6h、8h、10h、12h等,优选为0.5h~2h;反应温度为20℃~120℃,例如可以为20℃、25℃、30℃、35℃、40℃、50℃、60℃、80℃、100℃、120℃等,优选为70~90℃。
进一步地,步骤s40中,将所述化合物iv和酸加入到第四反应溶剂中进行反应后,通过加热回流溶清,冷却降温析晶,再干燥得到所述目标化合物。
析晶温度为0℃~40℃,例如可以为0℃、1℃、2℃、3℃、4℃、5℃、10℃、20℃、30℃、40℃等,优选为0~20℃;析晶时间为1h~48h,例如可以为1h、2h、3h、6h、12h、18h、24h、30h、36h、42h、48h等,优选为1h~3h;干燥温度可为20~100℃,例如可以为20℃、25℃、30℃、35℃、40℃、50℃、60℃、80℃、100℃、120℃等,优选为40~80℃;干燥时间为1h~72h,例如可以为1h、2h、3h、6h、12h、18h、24h、30h、36h、48h、60h、72h等,优选为4~8h。
通过本发明提供的吲哚衍生物的制备方法制备的目标化合物中,赛洛多辛中间体脱氢杂质的标定含量大于95.0%,甚至可以达到98%以上,可作为杂质标准品,应用于赛洛多辛原料及其制剂的杂质定性、定量研究,对有效控制赛洛多辛原料及其制剂的质量具有重要意义。
应当理解的是,吲哚衍生物可以包括赛洛多辛中间体杂质,还可以包括其他类型的化合物,此处不做限制。
本发明先后进行过多次试验,现举一部分试验结果作为参考对发明进行进一步详细描述,下面结合具体实施例进行详细说明。
实施例1
步骤s101:在500ml的三口瓶中,分别加入5.11g5-[(2r)-2-氨基丙基]-1-[3-(苯甲酰氧基)丙基]-2,3-二氢-7-氰基-1h-吲哚酒石酸盐、150ml二氯甲烷、5ml三乙胺,并进行搅拌;
搅拌溶解后,分批加入二叔丁基二碳酸酯2.52g,并在室温下搅拌反应2h,得到反应液;
向上述反应液中加入100ml纯化水,搅拌10min,然后静置分层;
分别用100ml纯化水和100ml饱和氯化钠洗涤静置分层后的有机相,然后用无水硫酸钠干燥有机相2h后,再过滤,得到滤液;
对滤液进行减压浓缩,得到3.84g淡黄色油状化合物ⅱ,化合物ⅱ为5-[(2r)-2-(叔丁氧羰基)氨基丙基]-1-[3-(苯甲酰氧基)丙基]-2,3-二氢-7-氰基-1h-吲哚,其收率为83.25%。
步骤s102:在250ml的三口瓶中,分别加入3.80g化合物ⅱ和40ml乙酸乙酯,并进行搅拌;
搅拌溶解后,加入3.27gddq,在室温下,反应过夜后得到反应液;
向上述反应液中加入30ml饱和碳酸氢钠,搅拌10min,然后静置分层;
用30ml饱和氯化钠洗涤静置分层后的有机相一次,然后用无水硫酸钠干燥有机相2h后,再过滤,得到滤液;
对滤液进行减压浓缩,得到黑色油状物;
柱层析纯化上述黑色油状物,并收集乙酸乙酯/石油醚=1/1的洗脱液,浓缩洗脱液,得到2.5g黄色油状化合物ⅲ5-[(2r)-2-(叔丁氧羰基)氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚,其收率为66.14%。
步骤s103:在250ml的三口瓶中,分别加入40ml的实施例3中的化合物ⅲ5-[(2r)-2-(叔丁氧羰基)氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚的二氯甲烷溶液和4ml三氟乙酸,在室温下,反应5h,并浓缩蒸干,得到残留物;
向上述残留物中加入50ml二氯甲烷,并用饱和碳酸氢钠洗涤两次,每次30ml;
用30ml饱和盐水洗涤有机相一次后,然后用无水硫酸钠干燥有机相2h后,再过滤,得到滤液;
对滤液进行减压浓缩,得到1.67g黄色油状化合物iv(5-[(2r)-2-氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚),其收率为85.20%。
步骤s104:在100ml的三口瓶中,分别加入1.20g化合物iv和20ml乙酸乙酯,并进行搅拌;
搅拌溶解后,加入0.30g乙二酸,升温回流并搅拌30min,然后冷却降温至15℃,搅拌1h,得到反应溶液;
过滤上述反应溶液,得到滤饼,用10ml乙酸乙酯淋洗过滤后得到的滤饼;然后在控温50℃的鼓风下,干燥淋洗后的滤饼6h,得到1.28g类白色固体(目标化合物,5-[(2r)-2-氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚草酸盐),其收率为85.33%,其纯度为98.15%(hplc)。
目标化合物的纯度检测方式为:以watersxbridge(4.6mm×150mm,5μm)为色谱柱,230nm为检测波长,0.01mol/l磷酸二氢钠溶液(调节ph值至6.0)-乙腈=75:25为流动相进行高效液相分析,得如图2所示的高效液相色谱图。
电喷雾质谱图如图3所示:esi-msm/z362.19[m+h]+,理论上5-[(2r)-2-氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚草酸盐游离碱分子式:c22h23n3o2,分子量:361.18。
核磁共振氢谱图如图4所示,具体化学位移见下:
1h-nmr(400mhz,dmso)δ7.91-7.89(d,2h),7.66(s,1h),7.65-7.61(m,2h),7.55(s,1h),7.49-7.47(m,2h),6.62(s,1h),4.67-4.65(m,2h),4.32-4.29(d,2h),3.49-3.45(m,1h),3.07-3.04(m,1h),2.83-2.80(m,1h),2.31-2.29(m,2h),1.13-1.12(d,3h)。
实施例2
步骤s201:在500ml的三口瓶中,分别加入5.15g5-[(2r)-2-氨基丙基]-1-[3-(苯甲酰氧基)丙基]-2,3-二氢-7-氰基-1h-吲哚酒石酸盐、150ml二氯甲烷、5ml三乙胺,并进行搅拌;
搅拌溶解后,分批加入二叔丁基二碳酸酯2.48g,并在室温下搅拌反应2h,得到反应液;
向上述反应液中加入100ml纯化水,搅拌10min,然后静置分层;
分别用100ml纯化水和100ml饱和氯化钠洗涤静置分层后的有机相,然后用无水硫酸钠干燥有机相2h后,再过滤,得到滤液;
对滤液进行减压浓缩,得到3.86g淡黄色油状化合物ⅱ,化合物ⅱ为5-[(2r)-2-(叔丁氧羰基)氨基丙基]-1-[3-(苯甲酰氧基)丙基]-2,3-二氢-7-氰基-1h-吲哚,其收率为83.01%。
步骤s202:在250ml的三口瓶中,分别加入3.80g化合物ⅱ和40ml二氯甲烷,并进行搅拌;
搅拌溶解后,加入3.85g二氧化锰,升温到40℃,反应过夜,得到反应液;
过滤上述反应液,滤除不溶物,得到状化合物ⅲ5-[(2r)-2-(叔丁氧羰基)氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚的二氯甲烷溶液。
步骤s203:在250ml的三口瓶中,分别加入40ml的化合物ⅲ5-[(2r)-2-(叔丁氧羰基)氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚的二氯甲烷溶液和4ml三氟乙酸,在室温下,反应5h,并浓缩蒸干,得到残留物;
向上述残留物中加入50ml二氯甲烷,并用饱和碳酸氢钠洗涤两次,每次30ml;
用30ml饱和盐水洗涤有机相一次后,然后用无水硫酸钠干燥有机相2h后,再过滤,得到滤液;
对滤液进行减压浓缩,得到1.95g黄色油状化合物iv(5-[(2r)-2-氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚),其收率为65.88%。
步骤s204:在100ml的三口瓶中,分别加入1.90g化合物iv和20ml乙酸乙酯,并进行搅拌;
搅拌溶解后,加入0.57g乙二酸,升温回流并搅拌30min,然后冷却降温至15℃,搅拌1h,得到反应溶液;
过滤上述反应溶液,得到滤饼,用10ml乙酸乙酯淋洗过滤后得到的滤饼;然后在控温50℃的鼓风下,干燥淋洗后的滤饼6h,得到2.05g类白色固体(目标化合物,5-[(2r)-2-氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚草酸盐),其收率为86.49%。
目标化合物的纯度检测方式为:以watersxbridge(4.6mm×150mm,5μm)为色谱柱,230nm为检测波长,0.01mol/l磷酸二氢钠溶液(调节ph值至6.0)-乙腈=75:25为流动相进行高效液相分析。
电喷雾质谱图和核磁共振氢谱图可参照实施例1。
实施例3
步骤s301:在500ml的三口瓶中,分别加入5.14g5-[(2r)-2-氨基丙基]-1-[3-(苯甲酰氧基)丙基]-2,3-二氢-7-氰基-1h-吲哚酒石酸盐、150ml二氯甲烷、5ml三乙胺,并进行搅拌;
搅拌溶解后,分批加入二叔丁基二碳酸酯2.18g,并在室温下搅拌反应2h,得到反应液;
向上述反应液中加入100ml纯化水,搅拌10min,然后静置分层;
分别用100ml纯化水和100ml饱和氯化钠洗涤静置分层后的有机相,然后用无水硫酸钠干燥有机相2h后,再过滤,得到滤液;
对滤液进行减压浓缩,得到3.92g淡黄色油状化合物ⅱ,化合物ⅱ为5-[(2r)-2-(叔丁氧羰基)氨基丙基]-1-[3-(苯甲酰氧基)丙基]-2,3-二氢-7-氰基-1h-吲哚,其收率为84.48%。
步骤s302:在250ml的三口瓶中,分别加入3.90g化合物ⅱ和40ml乙酸乙酯,并进行搅拌;
搅拌溶解后,加入3.25gddq,在室温下,反应过夜后得到反应液;
向上述反应液中加入30ml饱和碳酸氢钠,搅拌10min,然后静置分层;
用30ml饱和氯化钠洗涤静置分层后的有机相一次,然后用无水硫酸钠干燥有机相2h后,再过滤,得到滤液;
对滤液进行减压浓缩,得到黑色油状物;
柱层析纯化上述黑色油状物,并收集乙酸乙酯/石油醚=1/1的洗脱液,浓缩洗脱液,得到2.4g黄色油状化合物ⅲ5-[(2r)-2-(叔丁氧羰基)氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚,其收率为61.86%。
步骤s303:在250ml的三口瓶中,分别加入40ml化合物ⅲ5-[(2r)-2-(叔丁氧羰基)氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚的二氯甲烷溶液和4ml三氟乙酸,在室温下,反应5h,并浓缩蒸干,得到残留物;
向上述残留物中加入50ml二氯甲烷,并用饱和碳酸氢钠洗涤两次,每次30ml;
用30ml饱和盐水洗涤有机相一次后,然后用无水硫酸钠干燥有机相2h后,再过滤,得到滤液;
对滤液进行减压浓缩,得到1.57g黄色油状化合物iv(5-[(2r)-2-氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚),其收率为83.51%。
步骤s304:在100ml的三口瓶中,分别加入1.10g化合物iv和15ml异丙醇,并进行搅拌;
搅拌溶解后,加入0.28g乙二酸,升温回流并搅拌30min,然后冷却降温至15℃,搅拌1h,得到反应溶液;
过滤上述反应溶液,得到滤饼,用10ml乙酸乙酯淋洗过滤后得到的滤饼;然后在控温50℃的鼓风下,干燥淋洗后的滤饼6h,得到1.02g类白色固体(目标化合物,5-[(2r)-2-氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚草酸盐),其收率为74.45%,其纯度为98.09%。
目标化合物的纯度检测方式为:以watersxbridge(4.6mm×150mm,5μm)为色谱柱,230nm为检测波长,0.01mol/l磷酸二氢钠溶液(调节ph值至6.0)-乙腈=75:25为流动相进行高效液相分析,得如图5所示的高效液相色谱图。其中,目标化合物(5-[(2r)-2-氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚草酸盐)乙二酸分子式:c24h25n3o6,分子量:451.17。
目标化合物(5-[(2r)-2-氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚草酸盐)游离碱分子式:c22h23n3o2,分子量:361.18。
电喷雾质谱图和核磁共振氢谱图可参照实施例1。
实施例4
步骤s401:在500ml的三口瓶中,分别加入5.20g5-[(2r)-2-氨基丙基]-1-[3-(苯甲酰氧基)丙基]-2,3-二氢-7-氰基-1h-吲哚酒石酸盐、150ml二氯甲烷、5ml三乙胺,并进行搅拌;
搅拌溶解后,分批加入二叔丁基二碳酸酯2.58g,并在室温下搅拌反应2h,得到反应液;
向上述反应液中加入100ml纯化水,搅拌10min,然后静置分层;
分别用100ml纯化水和100ml饱和氯化钠洗涤静置分层后的有机相,然后用无水硫酸钠干燥有机相2h后,再过滤,得到滤液;
对滤液进行减压浓缩,得到3.84g淡黄色油状化合物ⅱ,化合物ⅱ为5-[(2r)-2-(叔丁氧羰基)氨基丙基]-1-[3-(苯甲酰氧基)丙基]-2,3-二氢-7-氰基-1h-吲哚,其收率为81.88%。
步骤s402:在250ml的三口瓶中,分别加入3.80g化合物ⅱ和40ml二氯甲烷,并进行搅拌;
搅拌溶解后,加入5.85g二氧化锰,升温到40℃,反应过夜,得到反应液;
过滤上述反应液,滤除不溶物,得到状化合物ⅲ5-[(2r)-2-(叔丁氧羰基)氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚的二氯甲烷溶液。
步骤s403:在250ml的三口瓶中,分别加入40ml化合物ⅲ5-[(2r)-2-(叔丁氧羰基)氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚的二氯甲烷溶液和4ml三氟乙酸,在室温下,反应5h,并浓缩蒸干,得到残留物;
向上述残留物中加入50ml二氯甲烷,并用饱和碳酸氢钠洗涤两次,每次30ml;
用30ml饱和盐水洗涤有机相一次后,然后用无水硫酸钠干燥有机相2h后,再过滤,得到滤液;
对滤液进行减压浓缩,得到1.89g黄色油状化合物iv(5-[(2r)-2-氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚),其收率为63.85%。
步骤s404:在100ml的三口瓶中,分别加入1.80g化合物iv和15ml异丙醇,并进行搅拌;
搅拌溶解后,加入0.45g乙二酸,升温回流并搅拌30min,然后冷却降温至15℃,搅拌1h,得到反应溶液;
过滤上述反应溶液,得到滤饼,用10ml乙酸乙酯淋洗过滤后得到的滤饼;然后在控温50℃的鼓风下,干燥淋洗后的滤饼6h,得到1.37g类白色固体(目标化合物,5-[(2r)-2-氨基丙基]-1-[3-(苯甲酰氧基)丙基]-7-氰基-1h-吲哚草酸盐),其收率为74.22%。
目标化合物的纯度检测方式为:以watersxbridge(4.6mm×150mm,5μm)为色谱柱,230nm为检测波长,0.01mol/l磷酸二氢钠溶液(调节ph值至6.0)-乙腈=75:25为流动相进行高效液相分析。
电喷雾质谱图和核磁共振氢谱图可参照实施例1。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!