春兰miR390a在控制植物根系发育中的应用的制作方法
本发明涉及植物基因工程技术领域,具体涉及一种春兰非编码rna(核糖核酸)mir390a在控制植物根系发育中的应用。
背景技术:
兰科(orchidaceae)是开花植物中最大科之一,全世界有25000多种,约占所有开花植物的10%。春兰(cymbidiumgoeringii)属于兰科兰属中的小花型地生兰种类,其花型奇特、花色淡雅,花香清幽,叶姿优美,观赏价值和经济价值极高。但春兰从幼苗到开花所需要的时间较长,野生春兰实生苗需十年左右方可开花,人工组培苗在养护得当的情况下也需至少三年才能开花。古语云:“根深叶茂。”根系发育良好,有助于地上其他营养器官的生长发育,进而令植物提前进入生殖发育期。因此,研究基因对植物根系发育的分子机制对春兰育种、生产及应用具有重要意义。而mir390在植物生长发育方面(尤其是根系)具有重要作用,可为植物的遗传改良提供重要依据。
microrna390s(mir390s)是植物micrornas中较为保守的古老家族之一,可参与多种植物生理过程,目前在其对植物生长发育方面的研究更多一些,包括植物侧生器官极性的形成、花器官形成及果实形成等。例如,油菜花的mir390参与了其早期胚胎的发育。osmir390前体基因的过表达可导致水稻出现顶端分生组织异常及营养阶段发育迟缓等表型缺陷。研究表明,mir390一般通过影响生长素信号来参与调控植物的生长发育过程。如,拟南芥atmir390能通过抑制arf3和arf4转录因子来延长幼年期推迟开花,从而间接调控花期。
根是植物生长所不可或缺的重要器官,顶端分生组织为控制其生长的关键部位,而根的顶端分生组织主要由静止中心、近端分生组织及伸长区域所组成。研究表明,mir390对植物的根系发育具有重要作用。在拟南芥根分生组织中的短期细胞扩增区域,arf5/monopteros通过结合mir390启动子上的auxre区域来直接调控其表达,进而影响根系的生长,而该过程对外源生长素有着强烈的响应。此外,拟南芥还可通过mir390/tas3/arf(arf2/arf3/arf4)途径来调控侧根的生长发育。然而,mir390如何对春兰根系的发育产生影响,目前尚未有研究进行说明,因此,利用基因工程技术,将从春兰中克隆获得mir390a前体基因转入其它植物中,对于研究其功能具有重要意义,同时极具应用前景。
技术实现要素:
本发明提供了一种春兰mir390a在控制植物根系发育中的应用。
一种春兰mir390a在控制植物根系发育中的应用,所述植物mir390a的核苷酸序列如seqidno.1或seqidno.2所示。
优选的,所述植物为春兰。
具体方法为:将包含所述春兰mir390a的前体基因连接到载体上,通过农杆菌介导转化到野生型拟南芥“哥伦比亚”,筛选,培养,获得转基因植株。
由于mirna本身很短,只有20bp左右,而前体基因相比较稍长一些,将其连接到载体上有利于拟南芥转化的实现,优选的,所述前体基因的序列如seqidno.3或seqidno.4所示。
本发明通过将春兰mir390a转化到野生型拟南芥“哥伦比亚”,经筛选培养获得t3代植株,发现过表达mir390a基因的拟南芥植株现幼苗期根系生长速度加快、真叶形成速度加快等表型,可见该基因在兰花及其它园艺植物生产、育种中将有广泛的用途。
附图说明
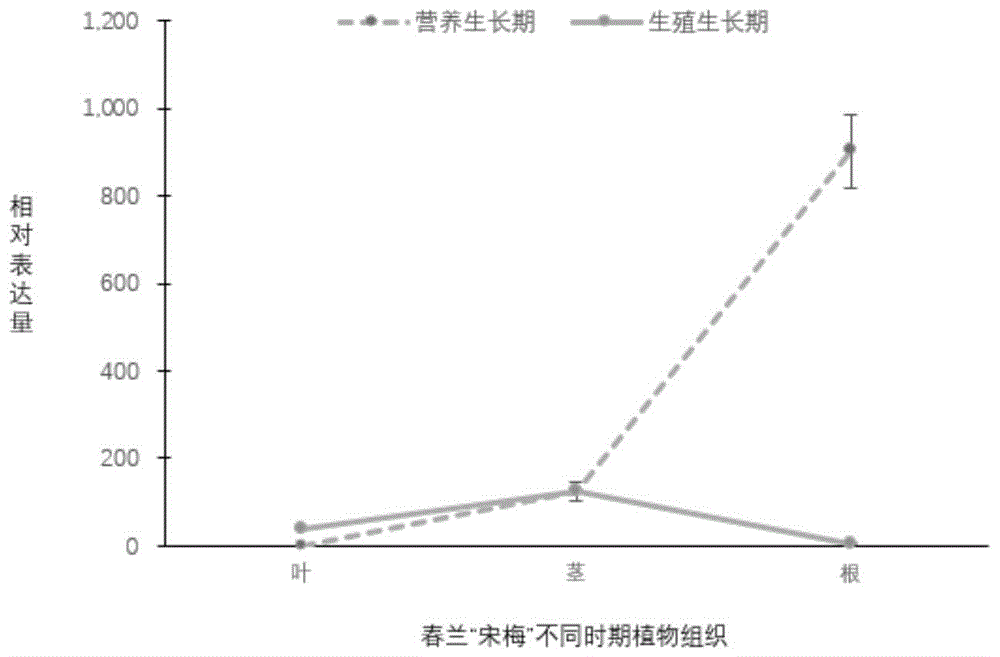
图1是春兰mir390a在春兰营养生长与生殖生长阶段各组织中的表达情况;
图2是pbi121和春兰mir390a基因前体片段编码基因(mir390a)重组载体示意图;
图3是mir390a过表达载体构建过程的凝胶图谱。a.mir390a前体pcr扩增凝胶图谱;b.pbi121-mir390a(mir390a前体)重组质粒菌液pcr扩增凝胶图谱;c.pbi121-mir390a(mir390a前体)重组质粒酶切验证图;
图4是t3代阳性转基因拟南芥植株鉴定结果。a.3个株系的mir390a前体pcr扩增凝胶图谱(m:dl2000marker;m1:阳性对照;m2:阴性对照;m3:空白对照);b.3个株系的mir390a荧光定量分析结果;
图5是播种至筛选培养基2周时,mir390a过表达拟南芥的表型(wt:野生型对照;35s:mir390a转基因植株)。
具体实施方式
下面结合具体实施例对本发明做进一步的说明。
实施例1基因的功能预测
本实施例所采用的材料是春兰“宋梅”营养生长时期及生殖生长时期的根、茎、叶,采后速冻于液氮中,超低温冰箱(-80℃)保存。
1)春兰各组织totalsmallrna的提取
按照takara植物总rna提取试剂盒的说明书进行,具体操作为:
将超低温冻存的春兰各组织迅速转移至用液氮预冷的研钵中,用研杵研磨组织,其间不断加入液氮,直至分别研磨成粉末状;将研磨成粉状的样品分别加入到含有450μlbufferpe的1.5ml灭菌tube中,用移液器反复吹打直至裂解液中无明显沉淀;将裂解液12,000rpm,4℃离心5分钟;将上清液小心吸取到新的1.5ml灭菌tube中。加入上清液1/10体积的buffernb,vortex振荡混匀,12,000rpm,4℃离心5钟;将上清液小心吸取到新的1.5ml灭菌tube中,加入450μl的bufferrl,使用移液枪将溶液混合均匀;加入混合液1.5倍体积的无水乙醇,使用移液枪将溶液混合均匀后,立即将混合液全部转入到rnaspincolumn中;12,000rpm,离心1分钟,弃滤液,将rnaspincolumn放回到2mlcollectiontube中;将600μl的80%乙醇加入至rnaspincolumn中,12,000rpm离心30秒钟,弃滤液;向rnaspincolumn膜中央加入50μldnasei反应液,室温静置15分钟;向rnaspincolumn膜中央加入350μl的bufferrwb,12,000rpm离心30秒钟,弃滤液;将600μl的80%乙醇加入至rnaspincolumn中,12,000rpm离心30秒钟,弃滤液;将rnaspincolumn重新安置于2mlcollectiontube上,12,000rpm离心2分钟;将rnaspincolumn安置于1.5ml的rnasefreecollectiontube上,在rnaspincolumn膜中央处加入30μl的rnasefreedh2o室温静置5分钟,12,000rpm离心2分钟洗脱rna。所得rna经浓度和纯度检测后存于-80℃冰箱保存备用。
吸取2μlrna利用1%琼脂糖凝胶电泳检测,结果显示28s和18s条带较为清晰,28s条带亮度约为18s的两倍,rna质量较好。通过微量核算蛋白测定仪检测rna纯度,od260/od280和od260/od230均在1.8~2.1之间,完整性较好,可用于反转录。
2)反转录与荧光定量分析
根据春兰microrna组学测序结果,利用primer5设计春兰mir390a基因茎环引物,参考序列为:5’-gtcgtatccagtgcagggtccgaggtattcgcactggatacgactggcgc-3’。同时,以18s作为内参基因,引物序列为:5’-tcgcagtggttcgtcttt-3’。以上述所提取的春兰各组织的totalsmallrna分别为模板,按照vazyme公司的hiscriptiii1ststrandcdnasynthesiskit试剂盒的说明书混合好体系,进行cdna反转录,反转录程序为:37℃15min,85℃5s。
将反转录产物稀释10倍,取1μl作模板,所用荧光定量引物如下:
mir390a-f:5’-cgcgaagctcaggagggata-3’
mir390a-r:5’-agtgcagggtccgaggtatt-3’
18s-f:5’-ggtcctattgtgttggct-3’
18s-r:5’-tcgcagtggttcgtcttt-3’
利用chamq™universalsybrqpcrmastermix试剂盒(vazyme公司)的说明书进行反应溶液的配制,在appliedbiosystems型实时荧光定量分析仪上运行pcr程序:95℃5min;95℃10s,60℃30s,循环40次;95℃15s,60℃1min,95℃15s。待反应结束,获得扩增曲线,通过steponesoftwarev2.3导出数据,利用excel进行数据分析,根据ct值用2-δδcq相对定量法计算相对表达量,数据分析结果如图1所示。
本实施例根据对荧光定量结果的分析,发现mir390a在春兰营养生长时期的根上表达量最高,说明其对植物营养生长期根系的生长发育有着重要作用。
实施例2基因的克隆与转化
本实施例所用的植物材料为春兰“宋梅”的新鲜叶片,拟南芥(arabidopsisthaliana)为“哥伦比亚(columbia)”型。所用的大肠杆菌菌株为trans5α,用于基因克隆与过表达载体构建,载体构建如图2所示;农杆菌菌株为gv3101,用于转化拟南芥;试验中所用植物表达载体为pbi121。所用菌株分别购自擎科生物科技有限公司和普利斯生物公司。
1)春兰叶片gdna的提取
按照takara植物genomicdna提取试剂盒的说明书进行,具体操作为:
将新鲜的春兰“宋梅”叶片转移至液氮预冷的研钵中,用研杵研磨组织,其间不断加入液氮,直至研磨成粉末状;迅速将研磨成粉状的样品加入到含有500μlbufferhsi和10μl50xdttbuffer混合液的1.5ml灭菌tube中混匀,向其中加入10μlrnasea,涡旋振荡混匀,于56℃金属浴温育10min。将62.5μl的bufferkac加入裂解好的样品中,用移液枪反复吹打混匀,将其在冰上放置5min,12,000rpm离心5分钟;小心吸取上清液,并转移至新的新的1.5ml灭菌tube中,加入与上清液等体积的buffergb并将二者混合均匀;将上述所得混合液分次移入安置在collectiontube的spincolumn中,12,000rpm离心1分钟,弃滤液;将500μlbufferwa加至spincolumn中,12,000rpm离心1分钟,弃滤液;将700μlbufferwb沿spincolumn管壁四周加入,12,000rpm离心1分钟,弃滤液;再次将700μlbufferwb沿spincolumn管壁四周加入,12,000rpm离心1分钟,弃滤液;12,000rpm离心2分钟,以确保无剩余液体残留在spincolumn中。将spincolumn安置于新的1.5ml灭菌tube中,向spincolumn膜的中央处加入30μl在65℃金属浴上温育过的无菌水,室温静置5min;12,000rpm离心2分钟以洗脱gdna。所得gdna经浓度和纯度检测后存于-80℃冰箱保存备用。
吸取2μlgdna利用1.5%琼脂糖凝胶电泳检测,结果显示仅有一条清晰的大分子条带,全基因组dna质量较好。通过微量核算蛋白测定仪检测gdna纯度,od260/od280和od260/od230均在1.8~2.1之间,,完整性较好,可用于pcr。
2)目的基因引物的设计及克隆
根据现有的春兰mirna组测序数据,利用其它物种的mir390相关基因序列进行blast同源比对,得到含春兰mir390a的前体序列。利用oligo6.0,prime5.0在春兰mir390a前体序列发卡结构两端设计相应引物,并在上加入所选酶切位点(xbai、smai)及pbi121上两酶切位点所在的同源臂,其引物序列为:
mir390a-xbai-f:5'-gagaacacgggggactctagaaatgtatgggagaaccattaaagct-3'(下划线部分为xbai酶切位点),
mir390a-smai-r:5'-ataagggactgaccacccgggagagcaggaagaaccaatagaactca-3'(下划线部分为smai酶切位点)。
以gdna为模板,利用primerstarmax高保真酶进行春兰mir390基因的克隆。pcr扩增体系(50μl)为:25μllprimerstarmax,2μlforwardprimer,2μlreverseprimer,2μltemplatedna,19μlddh2o。pcr程序为:反应条件为94℃预变性3min,98℃变性10s,60℃退火15s,72℃延伸30s,32个循环,72℃总延伸5min,16℃保温。
pcr反应完成后,取全部pcr产物通过1.8%琼脂糖凝胶电泳检测(pcr扩增结果如图3a)并切割目的片段,凝胶回收纯化pcr目的扩增产物。采用transgen公司的dna凝胶回收试剂盒,进行目的片段纯化回收,具体操作为:将单一目的条带从琼脂糖凝胶中切下,放入干净的离心管中,称取重量;向胶块中加入3倍体积溶液gsb(如果凝胶为0.1g,其体积可视为100μl,则加入300μlgsb溶液),55℃水浴放置,其间不断温和上下翻转离心管,直至胶块完全溶解;将融化的凝胶溶液降至室温,加入1倍体积异丙醇(如果凝胶为0.1g,则加入100μl异丙醇),轻柔混匀;将混合液加入离心柱中,室温放置1min,12000rpm离心1min,弃流出液,后将离心柱放回收集管中;向离心柱内加入650μl溶液wb,12000rpm离心1min,弃流出液;12000rpm离心2min,尽量除尽残留的wb,将吸附柱置于室温开盖放置5min,彻底晾干;将离心柱放到一个干净离心管中,向吸附膜中间位置悬空滴加30μlddh2o(ddh2o需提前置于60~70℃水浴预热),室温静置2min,12000rpm离心2min收集dna溶液。取2μl回收纯化后的产物,使用1.5%琼脂糖进行凝胶电泳检测,其余放置在-20℃冰箱,后续将用于与pbi121载体连接,构建过表达载体。
3)质粒的提取:
按照天根质粒小提中量试剂盒说明书提取质粒,具体步骤如下:
取过夜培养的10ml菌液,12000rpm离心1min,去掉上清液;取500μlp1溶液(含rnasea)加至留有菌体沉淀的离心管中,使用涡旋仪彻底悬浮菌体沉淀;取500μlp2溶液加至离心管中,温和上下翻转时菌体裂解充分,取700μlp3溶液加至离心管中,立即温和上下翻转,充分混匀,当出现白色絮状沉淀后,12000rpm离心10min;取500μl平衡液bl加至吸附柱cp4中,12000rpm离心1min,弃掉收集管中的废液,将吸附柱放回收集管,将收集的上清液分批加入过滤柱cs中,12000rpm离心2min,小心将收集管中收集的溶液分批加入吸附柱cp4中,12000rpm离心1min,弃掉收集管中的废液,将吸附柱cp4放回收集管;取500μl去蛋白液pd加至吸附柱cp4中,12000rpm离心1min,弃掉收集管中废液,将吸附柱cp4重新放回收集管;取600μl漂洗液pw(含无水乙醇)加至吸附柱cp4中,12000rpm离心1min,弃掉收集管中的废液,将吸附柱cp4放回收集管,12000rpm离心2min,去除吸附柱中残余的漂洗液;将吸附柱cp4移至新的1.5ml离心管中,向吸附膜中间加入60μlddh2o;室温静置2min,12000rpm离心1min,离心管中收集的溶液即为质粒。最后测定质粒浓度,为下一步实验做准备。
4)双酶切反应
将提取的pbi121质粒用xbai和smai在37℃条件下酶切30min,电泳回收线性载体,-20℃保存备用。双酶切反应体系为50μl:pbi121质粒20μl,5×buffer5μl,xbai1μl,smai1μl,ddh2o23μl。
5)重组反应
琼脂糖凝胶电泳检测酶切后所回收到的目的基因和载体pbi121,根据所检测出的纯度和浓度,按连接体系加入各试剂。其中,目的片段分子数:载体分子数=3:1~5:1,连接反应体系为:线性化pbi121载体7μl,插入片段3μl,5×ceiibuffer4μl,exnaseii2μl,ddh2oupto20μl。在37℃下反应30min,常温放置(勿立即置于冷却),10min后转化至大肠杆菌感受态。
6)连接产物转入大肠杆菌
从超低温冰箱中取出感受态细胞trans5α菌株,置于冰上融化。吸取10μl重组产物加入到100μl感受态细胞中;将离心管置于冰上冰浴10min;42℃水浴锅中水浴,热激90s,期间不要摇动;后立即置于冰上冰浴2min;在超净台中加入500μl无抗生素的液体培养基,37℃、200rpm摇25min复苏;6000rpm离心1min,吸去350μl上清;将沉淀的菌体重悬,涂布于lb平板(kana的浓度为50mg/l),37℃培养过夜。
7)重组子的鉴定
挑取平板上的单菌落接种到含有抗生素(kana)的lb液体培养基中,37℃、200rpm震荡培养过夜。使用目的基因全长引物进行菌液pcr,以筛选阳性克隆,菌检结果如图3b所示。筛选后的阳性克隆送南京思普金公司测序。测序结果正确的阳性克隆,扩大培养后,使用天根质粒提取试剂盒提取质粒并进行双酶切验证,判断酶切后片段大小是否一致,酶切结果如图3c所示,其中,m:dl2000marker;1:mir390a前体与pbi121连接后用xbai和smai双酶切。
8)农杆菌感受态细胞的制备与转化
本实施例利用农杆菌gv3101来制备农杆菌感受态,进行拟南芥的侵染实验;农杆菌感受态制备过程为:挑取已经活化好的农杆菌单菌落,接种于5ml液体lb培养基中,28℃、250rpm摇菌培养20-24h;吸取2ml菌液,接种到含有50ml液体lb培养基的三角瓶中,28℃、250rpm摇菌至od600值为0.8左右;将扩大繁殖后的菌液置于冰上冰浴30min,4℃、5000rpm离心5min,弃上清;加入10ml经预冷的0.1mo1/lcacl2溶液,充分悬浮沉淀的菌体;4℃,5000rpm离心5min,弃去上清;加入1ml预冷的20mmo1/lcacl2溶液充分悬浮菌体,即得到所要制备的gv3101感受态细胞,用离心管将其分装成100µl/管,迅速加入20%的无菌甘油,-80℃放置保存。
重组子的农杆菌转化:冰浴,使农杆菌感受态细胞融化,将600ng经回收纯化后的质粒加入到100μl的农杆菌感受态中,轻轻混匀,冰浴5min;使用液氮速冻5min,37℃金属浴中热激5min,迅速置于冰上冰浴5min;加入800μl不含任何抗生素的lb培养基,28℃,200rpm复苏2h;4000rpm离心3min,吸掉部分液体培养基;使用移液枪充分混匀剩余的菌液,后涂布于添加50mg/l卡那霉素和100mg/l的庆大霉素(gv3101)的固体lb培养基上;28℃倒置培养30~48h。
农杆菌重组子的鉴定:从平板培养基上挑取长出的单菌落,接种于含有相应抗生素的液体培养基中;28℃,200rpm培养过夜;使用35s-f分别搭配如下引物进行菌液pcr,引物序列为:
35s-f:5'-gatagtggaaaaggaaggtg-3',
35s:mir390a-r:5'-ataagggactgaccacccgggagagcaggaagaaccaatagaactca-3'。
pcr产物经1.5%琼脂糖凝胶电泳检测,鉴定是否含有目的片段,鉴定后的阳性克隆加入适量无菌50%甘油,于-80℃保存备用。
9)农杆菌介导的拟南芥的转化
采用花序侵染法将目的基因转入拟南芥中,具体操作方法为:拟南芥(col野生型)保持健康生长状态至开花;活化携带有目的基因的农杆菌gv3101菌株。挑取单菌落,接种于5ml含有卡那霉素和庆大霉素的lb培养基上,28℃、200rpm摇菌至菌液刚刚变浑浊,约8-10h;吸取1ml菌液,接种到三角瓶中(50ml)摇菌24h,至od值约为0.8左右;将菌液6000rpm在室温下离心5min,去除上清后收集菌体,用ph5.8的3%蔗糖溶液悬浮;浸泡前,加入silwetl-77,浓度为0.03%(300μl/l),晃出泡沫;将拟南芥的地上部分在农杆菌悬浮溶液中浸泡1min,期间轻轻晃动;将浸过的拟南芥平躺在托盘中,用保鲜膜覆盖,锡箔纸密封避光,放置24h;揭开锡箔纸,正长条件下培养,当种子成熟时停止浇水。
3%蔗糖溶液重悬液各成分如下:ms培养基,添加蔗糖30g/l,silwet-77300µl/l。(注意:配制后ph调制5.8,菌液离心重悬后再加入silwetl-77;重悬液和菌液的换算关系为:重悬液用量:菌液od*菌液体积=0.8*重悬液)。
10)转基因植株的筛选
收集的t1代转基因拟南芥的种子,用酒精和次氯酸钠进行灭菌,步骤为:取适量获得的转基因种子放置于1.5ml离心管中,用0.8%naclo与乙醇混合液(现配,比例为体积比1:1)浸泡5min;75%酒精灭菌5~6次,每次2min;无菌水冲洗3~4次;用0.1%琼脂糖溶液悬浮。
将灭菌后的转基因拟南芥种子播种于含有抗生素(卡那霉素50mg/l和头孢霉素100mg/l)的ms固体培养基上,锡纸包裹放入4℃冰箱中春化。2天后从冰箱中取出,将培养基置于22℃,光照培养。大约一周后将培养基上可以正常生长的拟南芥移植与土中,继续生长。待植株长至一定程度,则取其叶片进行dna检测以获得阳性植株。
本实施例克隆得到1个春兰mir390a前体基因,命名为mir390a,其核苷酸序列如seqidno.3或seqidno.4所示,mir390a基因核苷酸序列长度为150bp,含有1个茎环结构,并在其臂上存在一对mir390a正/反义互补序列,该mir390a成熟体核苷酸序列如seqidno.1或seqidno.2所示。随后,将已连有春兰mir390a前体基因的35s:mir390a过表达重组载体转入模式植物拟南芥中,并获得了t1代阳性植株。
实施例3春兰mir390a控制植物根系发育的功能鉴定
1)转基因纯合植株的获得:收获的转基因t1代种子经灭菌,筛选培养后,再移植于营养土中,22℃,16h光照/8h黑暗培养;经检测后保留初步确认的转基因植株,待成熟后收获t1代种子,进行编号,得到t2代;同t1代一样,将t2代种子经灭菌后涂布于含抗生素的筛选培养基上,置于22℃,连续光照;10天左右,对不同编号的t2代种子进行存活率统计,选取存活比例为75%的植株移植与营养土中按照22℃,16h光照/8h黑暗培养,并取叶片进行阳性检测;阳性t2代植株继续进行编号,收集种子,得到t3代种子;将种子灭菌后,用筛选培养基筛选,置于光下连续光照培养;10天左右,观察不同编号的t3代植株,全部存活并且没有出现分离的为t3代纯合植株。
2)转基因植株的dna检测
取适量t3代拟南芥和转基因植株的幼嫩叶片,采用ctab法提取dna,具体操作步骤为:将适量叶片置于灭菌处理后的2ml离心管中,加入700µl的ctab溶液,用球磨仪彻底研磨,65℃静置10min;加入等体积的氯仿:异戊醇,数次颠倒使其混合均匀,14000rpm离心10min;将上清移至新的无菌离心管中,加入等体积的异丙醇,颠倒混匀数次,室温静置2min,14000rpm离心10min,倒掉上清;加入70%的无水乙醇,使用移液枪吹打洗涤两次,14000rpm离心1min,弃去上清;吹干表面液体,加入20µlddh2o溶解。取上述提取的转基因和野生型拟南芥的dna,用mir390a基因的特异性引物进行pcr检测。
春兰mir390a基因转化拟南芥至t3代,共获得3个过表达mir390a基因拟南芥株系。以重组质粒为阳性对照,以野生型为阴性对照,以水为空白对照,pcr结果如图4a所示。
3)转基因植株的荧光定量pcr检测
从上述3个过表达春兰mir390a基因拟南芥株系的幼嫩茎生叶中提取总smallrna,反转录及荧光定量引物、方法及程序同实施例1。以u6作为内参基因,其荧光定量引物如下,其反转录引物为以下u6-r。
u6-f:5’-ggtgctaagaagaggaagaat-3’
u6-r:5’-ctccttctttctggtaaacgt-3’
反应结束后,数据分析方式同实施例1,根据ct值用2-δδcq相对定量法计算相对表达量,最终数据分析结果如图4b所示。
4)t3代纯和转基因植株表型观察
选取其中表型明显的转基因植株进行观察,结果如图5所示,相比于野生型,转基因拟南芥植株在幼苗期生长较快,在种子拨入筛选培养基第2周,其根系长度约有1cm,而野生型的根系则约为0.2cm。此外,幼苗期转基因植株的地上部分生长速度明显快于野生型,第2周时其真叶已达4片以上,而野生型真叶仅长出2片。
本实施例将转春兰35s:mir390a拟南芥植株筛选、培养并获得t3代纯合植株,进行表型观察。从结果可以看出,过表达35s:mir390a的拟南芥t3代纯合植株出现幼苗期根系生长速度加快、真叶形成速度加快等表型。
序列表
<110>南京林业大学
<120>春兰mir390a在控制植物根系发育中的应用
<160>14
<170>siposequencelisting1.0
<210>1
<211>22
<212>rna
<213>春兰cymbidiumgoeringii
<400>1
aagcucaggagggauagcgcca22
<210>2
<211>22
<212>dna
<213>春兰cymbidiumgoeringii
<400>2
aagctcaggagggatagcgcca22
<210>3
<211>150
<212>rna
<213>春兰cymbidiumgoeringii
<400>3
aauguaugggagaaccauuaaagcucaggagggauagcgccaugaaugauuaugcaugua60
aaaccuggugaugagcuugcucucuuuaucagguuuuuuguuucauucugugacgcuauc120
uauccugaguucuauugguucuuccugcuu150
<210>4
<211>150
<212>dna
<213>春兰cymbidiumgoeringii
<400>4
aatgtatgggagaaccattaaagctcaggagggatagcgccatgaatgattatgcatgta60
aaacctggtgatgagcttgctctctttatcaggttttttgtttcattctgtgacgctatc120
tatcctgagttctattggttcttcctgctt150
<210>5
<211>20
<212>dna
<213>人工序列(artiartificialsequence)
<400>5
cgcgaagctcaggagggata20
<210>6
<211>20
<212>dna
<213>人工序列(artiartificialsequence)
<400>6
agtgcagggtccgaggtatt20
<210>7
<211>18
<212>dna
<213>人工序列(artiartificialsequence)
<400>7
ggtcctattgtgttggct18
<210>8
<211>18
<212>dna
<213>人工序列(artiartificialsequence)
<400>8
tcgcagtggttcgtcttt18
<210>9
<211>46
<212>dna
<213>人工序列(artiartificialsequence)
<400>9
gagaacacgggggactctagaaatgtatgggagaaccattaaagct46
<210>10
<211>47
<212>dna
<213>人工序列(artiartificialsequence)
<400>10
ataagggactgaccacccgggagagcaggaagaaccaatagaactca47
<210>11
<211>20
<212>dna
<213>人工序列(artiartificialsequence)
<400>11
gatagtggaaaaggaaggtg20
<210>12
<211>47
<212>dna
<213>人工序列(artiartificialsequence)
<400>12
ataagggactgaccacccgggagagcaggaagaaccaatagaactca47
<210>13
<211>21
<212>dna
<213>人工序列(artiartificialsequence)
<400>13
ggtgctaagaagaggaagaat21
<210>14
<211>21
<212>dna
<213>人工序列(artiartificialsequence)
<400>14
ctccttctttctggtaaacgt21
- 还没有人留言评论。精彩留言会获得点赞!