一种单氰基取代的多羧酸有机配体及其制备方法
1.本发明属于有机化工合成技术领域,具体涉及一种单氰基取代的多羧酸有机配体及其制备方法。
背景技术:
2.近年来,功能化的金属有机骨架材料(mofs)因为有特定的官能团,在气体吸附、重金属离子吸附等方面有着独特的应用引起了人们极大的关注。在有机配体上直接合成出目标官能团,并进一步合成功能化有机金属骨架材料也一直是科研工作者关注的热点。鉴于金属有机骨架材料固有的较大的比表面积与多孔性,把含有氰基的官能团直接引入到有机配体中,进而可以得到氰基功能化的金属有机骨架材料,该材料在气体吸附和重金属离子吸附方面有着较好的应用。mofs材料的合成离不开有机配体,如何合成氰基功能化的多羧酸有机配体迫在眉睫。通过查阅大量文献,发现迄今为止没有合成出2-氰基-4,4'-联苯二甲酸这种含有氰基的多羧酸配体,本发明设计了一条可行的路线并成功得到该羧酸配体。
技术实现要素:
3.本发明是为了解决上述问题而进行的,目的在于提供一种单氰基取代的多羧酸有机配体及其制备方法。
4.本发明提供了一种单氰基取代的多羧酸有机配体,其化学名称为-氰基-4,4'-联苯二甲酸,结构式如下:
5.本发明还提供了一种单氰基取代的多羧酸有机配体的制备方法,其特征在于,包括以下步骤:步骤1,以联苯二甲酸二甲酯为原料,经过硝化反应得到2-硝基-4,4'-联苯二甲酸二甲酯;步骤2,采用2-硝基-4,4'-联苯二甲酸二甲酯经过还原反应得到2-氨基-4,4'-联苯二甲酸二甲酯;步骤3,采用2-氨基-4,4'-联苯二甲酸二甲酯经过重氮化反应后再经过碘化反应,得到2-碘-4,4'-联苯二甲酸二甲酯;步骤4,采用2-碘-4,4'-联苯二甲酸二甲酯经过氰化反应得到2-氰基-4,4'-联苯二甲酸二甲酯;步骤5,采用2-氰基-4,4'-联苯二甲酸二甲酯经过水解得到2-氰基-4,4'-联苯二甲酸。
6.在本发明提供的单氰基取代的多羧酸有机配体的制备方法中,还可以具有这样的特征:其中,步骤1中,硝化反应中的硝化剂为硝酸。
7.在本发明提供的单氰基取代的多羧酸有机配体的制备方法中,还可以具有这样的
特征:其中,步骤2中,还原反应中的还原剂为锡。
8.在本发明提供的单氰基取代的多羧酸有机配体的制备方法中,还可以具有这样的特征:其中,步骤3中,碘化反应中的碘化剂为碘化钾或碘化钠。
9.在本发明提供的单氰基取代的多羧酸有机配体的制备方法中,还可以具有这样的特征:其中,步骤4中,氰化反应中的氰化剂为氰化亚铜。
10.在本发明提供的单氰基取代的多羧酸有机配体的制备方法中,还可以具有这样的特征:其中,步骤4中,采用氰化亚铜与2-碘-4,4'-联苯二甲酸二甲酯通过微波反应得到2-氰基-4,4'-联苯二甲酸二甲酯。
11.本发明还提供了一种单氰基取代的多羧酸有机配体在制备金属骨架材料中的应用。
12.发明的作用与效果
13.本发明以联苯二甲酸二甲酯为原料,经过硝化反应、还原反应、重氮化反应、碘化反应、氰化反应及水解反应得到单氰基取代的多羧酸有机配体即2-氰基-4,4'-联苯二甲酸。本发明提供的合成路线反应简单,普遍适用,通过氰基的引入,使得以后用该法合成金属有机骨架材料的种类得到丰富。
14.另外,每一步反应收率都很高,水解反应的产率高达97%。
15.此外,反应条件温和,所需温度为0℃~100℃,反应不需要无水无氧,操作十分方便,分离提纯只需层析分离纯化或抽滤洗涤即可,简单易行。
16.此外,所采用的原料与溶剂价格便宜,易于得到,均为分析纯,且不需要任何后处理即可使用。
17.另外,利用本发明合成出来的2-氰基-4,4'-联苯二甲酸,可以进一步合成出氰基功能化的金属有机骨架材料。而该材料有着较大的比表面积和孔径,在气体吸附方面有着很大的应用,并且该材料的氰基官能团可以通过后合成修饰法进一步反应,形成其他功能化的金属有机骨架材料。
附图说明
18.图1是本发明的实施例中化合物a的核磁氢谱;
19.图2是本发明的实施例中化合物b的核磁氢谱;
20.图3是本发明的实施例中化合物c的核磁氢谱;
21.图4是本发明的实施例中化合物d的核磁氢谱;
22.图5是本发明的实施例中化合物l的核磁氢谱;以及
23.图6是本发明的实施例中化合物l的核磁碳谱。
具体实施方式
24.为了使本发明实现的技术手段、创作特征、达成目的与功效易于明白了解,以下结合实施例及附图对本发明一种单氰基取代的多羧酸有机配体及其制备方法作具体阐述。
25.本发明的实施例中所采用的原料与溶剂均为分析纯,通过一般商业途径购买可以得到。
26.本发明中的实施例中实验室配制的混酸为950μl~1500μl浓度为50%~70%的浓
硝酸与1ml~10ml的浓硫酸混合而成。
27.本发明提供的单氰基取代的多羧酸有机配体即2-氰基-4,4'-联苯二甲酸的合成路线如下:
[0028][0029]
<实施例>
[0030]
本实施例具体阐述2-氰基-4,4'-联苯二甲酸的合成过程。
[0031]
步骤1,以联苯二甲酸二甲酯为原料,经过硝化反应得到2-硝基-4,4'-联苯二甲酸二甲酯。
[0032]
具体操作为:称取4,4'-联苯二甲酸二甲酯(4.00g,14.8mmol)于单口瓶中,加入10ml~100ml浓硫酸,室温搅拌全溶后,置于0℃~10℃水浴中,缓慢的逐滴加入混酸(69%浓硝酸964μl与6ml浓硫酸的混合溶液),滴加过程中溶液温度应始终低于5℃,并在冰水浴中反应10min~60min,反应结束后倒入200ml~500ml冰水中,析出大量白色固体,用300ml~500ml乙酸乙酯(或二氯甲烷)萃取3次,有机相用300ml~500ml饱和食盐水洗涤至ph为中性,并用无水硫酸镁或无水硫酸钠干燥5min~30min,减压除去有机溶剂,利用硅胶色谱柱层析分离纯化,展开剂为石油醚:乙酸乙酯(10:1),得到淡黄色粉末2-硝基-4,4'-联苯二甲酸二甲酯4.18g,即化合物a,产率为89.7%。对得到的化合物a进行核磁检测,检测结果见图1。
[0033]
图1是本发明的实施例中化合物a的核磁氢谱。
[0034]
由图1可知,1h nmr(600mhz,cdcl3)δ8.56(s,1h),8.29(d,j=8.0hz,1h),8.12(d,j=8.4hz,2h),7.54(d,j=7.8hz,1h),7.40(d,j=8.4hz,2h),4.00(s,3h),3.95(s,3h).
[0035]
h的总数目和理论上合成配体的h的总数目一致,都为13个h,8.56m(1h)的单峰、8.29ppm(1h)的双峰、8.12ppm(2h)的双峰、7.54ppm(1h)的双峰、7.40ppm(2h)的双峰分别归属于苯环上7个位置的氢信号,4.00ppm(3h)的单峰、3.95ppm(3h)的单峰分别归属于酯基上两个甲基位置的氢信号。由以上可知,化合物a即为2-硝基-4,4'-联苯二甲酸二甲酯。
[0036]
步骤2,2-硝基-4,4'-联苯二甲酸二甲酯经过还原反应得到2-氨基-4,4'-联苯二甲酸二甲酯。
[0037]
具体操作为:称取2-硝基-4,4'-联苯二甲酸二甲酯(2.00g,6.3mmol)于双口瓶中,再加入100ml~500ml甲醇(或乙醇),搅拌后加入锡粉0.775g~5.000g(在本实施例中,加入的锡粉为4.40g,36.7mmol),然后逐滴加入浓度为0.5mol/l~3.0mol/l的稀盐酸溶液60ml
~100ml(在本实施例中,加入60ml 2mol/l稀盐酸溶液),升温至700℃~100℃,在回流状态下反应3h~10h,溶液由浑浊变成透明澄清,反应结束后冷却至室温,倒入200ml~500ml冰水中,产生大量沉淀,并用naoh或者koh溶液(0.1mol/l~5mol/l)调节溶液ph至7-10(在本实施例中,用2mol/l的naoh溶液调节溶液ph至10),然后用200ml~500ml乙酸乙酯(或二氯甲烷)萃取三次,再用200ml~500ml饱和食盐水洗涤一次,有机相用无水硫酸镁或无水硫酸钠干燥5min~30min,减压除去有机溶剂,利用硅胶色谱柱层析分离纯化,展开剂为石油醚:乙酸乙酯(10:1),得到淡黄色粉末2-氨基-4,4'-联苯二甲酸二甲酯1.02g,即化合物b,产率为56.3%。对得到的化合物b进行核磁检测,检测结果见图2。
[0038]
图2是本发明的实施例中化合物b的核磁氢谱。
[0039]
由图2可知,1h nmr(600mhz,dmso)δ7.96(d,j=7.8hz,2h),7.54(d,j=7.8hz,2h),7.36(s,1h),7.15(d,j=7.8hz,1h),7.06(d,j=7.8hz,1h),5.19(s,2h),3.81(s,3h),3.76(s,3h).
[0040]
h的总数目和理论上合成配体的h的总数目一致,都为13个h,7.96m(2h)的双峰、7.54ppm(2h)的双峰、7.36ppm(1h)的单峰、7.15ppm(1h)的双峰、7.06ppm(1h)的双峰分别归属于苯环上7个位置的氢信号,5.19ppm(2h)的单峰分别归属于苯环上氨基的氢信号,3.81ppm(3h)的单峰、3.76ppm(3h)的单峰分别归属于酯基上两个甲基位置的氢信号。由以上可知,化合物b即为2-氨基-4,4'-联苯二甲酸二甲酯。
[0041]
步骤3,2-氨基-4,4'-联苯二甲酸二甲酯经过重氮化反应后再经过碘化反应,得到2-碘-4,4'-联苯二甲酸二甲酯。
[0042]
具体操作为:称取2-氨基-4,4'-联苯二甲酸二甲酯(1.42g,5.0mmol)于单口瓶中,加入5ml 10%~30%的稀盐酸、稀硫酸或者稀硝酸(在本实施例中,加入5ml 15%的稀盐酸),反应体系置于-5℃~10℃并继续搅拌5min~15min,逐滴加入nano2溶液(由0.40g~0.70g,5.8mmol~10.0mmol的nano2溶于1ml~5ml蒸馏水配制而成,在本实施例中,用nano2(0.40g,5.8mmol)溶于3ml蒸馏水配制而成),继续反应5min~30min后,向体系加入碘化物(碘化钾或碘化钠)6mmol~10mmol(在本实施例中,加入ki 0.996g,6mmol),升温至30℃~100℃,继续反应0.5h~3h后,向体系加入30ml~100ml蒸馏水,冷却至室温,用100ml乙酸乙酯(或二氯甲烷)萃取三次,然后依次用100ml~200ml,10%~50%的na2so3溶液洗涤一次(在本实施例中,用100ml 10%na2so3溶液洗涤一次),100ml~200ml饱和食盐水洗涤一次,有机相再用无水硫酸镁或无水硫酸钠干燥5min~30min,减压蒸馏除去有机溶剂,利用硅胶色谱柱层析分离纯化,展开剂为石油醚:乙酸乙酯(10:1),得到淡黄色粉末2-碘-4,4'-联苯二甲酸二甲酯1.48g,即化合物c,产率为74.0%。对得到的化合物c进行核磁检测,检测结果见图3。
[0043]
图3是本发明的实施例中化合物c的核磁氢谱。
[0044]
由图3可知,1h nmr(600mhz,cdcl3)δ8.62(s,1h),8.12(d,j=7.8hz,2h),8.06(d,j=7.8hz,1h),7.42(d,j=7.8hz,2h),7.36(d,j=7.8hz,1h),3.95(s,6h).
[0045]
h的总数目和理论上合成配体的h的总数目一致,都为13个h,8.62m(1h)的单峰、8.12ppm(2h)的双峰、8.06ppm(1h)的双峰、7.42ppm(2h)的双峰、7.36ppm(1h)的双峰分别归属于苯环上7个位置的氢信号,3.95ppm(6h)的单峰归属于酯基两个甲基位置的氢信号。由以上可知,化合物c即为2-碘-4,4'-联苯二甲酸二甲酯。
[0046]
步骤4,2-碘-4,4'-联苯二甲酸二甲酯经过氰化反应得到2-氰基-4,4'-联苯二甲酸二甲酯。
[0047]
具体操作为:分别称取2-碘-4,4'-联苯二甲酸二甲酯(0.40g,1.0mmol)、1.0mmol~10mmol氰化物(cucn、氰化钾或氰化锌中的任意一种,在本实施例中,加入cucn 0.12g,1.3mmol)于微波管中,加入15mlnmp(氮甲基吡咯烷酮)或dmf(n,n-二甲基甲酰胺),超声5min~30min后,放入微波反应器中,3min~20min升温至100℃~180℃,并反应0.5h~20h,冷却至室温倒入100ml~200ml的浓度为5%-25%的氨水溶液(在本实施例中,用100ml 15%的氨水溶液)中,搅拌5min~30min,过滤得到粗产物固体,用100ml乙酸乙酯或二氯甲烷溶解,并用100ml饱和食盐水清洗一次,有机相用无水硫酸镁或无水硫酸钠干燥5min~30min,减压除去有机溶剂,利用硅胶色谱柱层析分离纯化,展开剂为石油醚:乙酸乙酯(10:1),得到白色粉末2-氰基-4,4'-联苯二甲酸二甲酯0.25g,即化合物d,产率为85.1%。对得到的化合物d进行核磁检测,检测结果见图4。
[0048]
图4是本发明的实施例中化合物d的核磁氢谱。
[0049]
由图4可知,1hnmr(600mhz,cdcl3)δ8.46(s,1h),8.31(d,j=9.0hz,1h),8.19(d,j=8.4hz,2h),7.66(d,j=7.8hz,2h),7.63(d,j=7.8hz,1h),3.99(s,3h),3.96(s,3h).
[0050]
h的总数目和理论上合成配体的h的总数目一致,都为13个h,8.46m(1h)的单峰、8.31ppm(1h)的双峰、8.19ppm(2h)的双峰、7.66ppm(2h)的双峰、7.63ppm(1h)的双峰分别归属于苯环上7个位置的氢信号,3.99ppm(3h)的单峰、3.96ppm(3h)的单峰分别归属于酯基上两个甲基位置的氢信号。由以上可知,化合物d即为2-氰基-4,4'-联苯二甲酸二甲酯。
[0051]
步骤5,2-氰基-4,4'-联苯二甲酸二甲酯经过水解得到2-氰基-4,4'-联苯二甲酸。
[0052]
具体操作为:称取2-氰基-4,4'-联苯二甲酸二甲酯(0.12g,0.4mmol)于单口瓶,加入5ml~50ml甲醇,在室温条件下搅拌,逐滴滴加1.0ml~5.0ml浓度为1mol/l~5mol/l的naoh或者koh溶液(在本实施例中,滴加1.1ml 1mol/l naoh溶液),保持室温(20℃~30℃)反应10h~24h,减压除去有机溶剂,加入10ml~50ml蒸馏水,置于0℃~10℃水浴中,逐滴加入浓度为5%~35%的稀盐酸、稀硫酸或稀硝酸(在本实施例中,加入15%的稀盐酸溶液),调节溶液ph至7~1,继续搅拌5min~35min,抽滤得到白色粉末,并用30ml蒸馏水与30ml正己烷各洗涤三次,烘干即可得到2-氰基-4,4'-联苯二甲酸0.10g,即化合物l,产率为97.0%。
[0053]
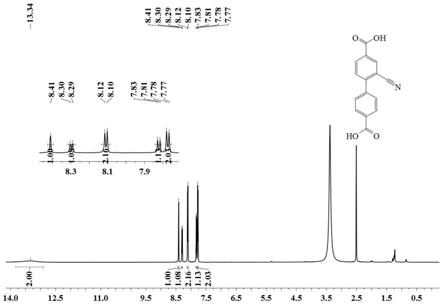
对得到的目标产物2-氰基-4,4'-联苯二甲酸,即化合物l进行核磁检测,检测结果见图5和图6。
[0054]
图5是本发明的实施例中化合物l的核磁氢谱。图6是本发明的实施例中化合物l的核磁碳谱。
[0055]
由图5可知,1h nmr(600mhz,dmso),δ13.34(s,2h),8.41(s,1h),8.29(d,j=7.8hz,1h),8.10(d,j=8.4hz,2h),7.82(d,j=8.4hz,1h),7.77(d,j=7.8hz,2h)。
[0056]
h的总数目和理论上合成配体的h的总数目一致,都为9个h,其中δ=13.34ppm(2h)处的单峰为苯环上羧酸氢的特征吸收峰,因羧酸氢为活泼氢,导致氢谱上有时无法检测到该活泼氢的吸收峰,8.41m(1h)的单峰、8.29ppm(1h)的双峰、8.10ppm(2h)的双峰、7.82ppm(1h)的双峰、7.77ppm(2h)的双峰分别归属于苯环上7个位置的氢信号。
[0057]
由图6可知,
13
c nmr(151mhz,dmso),δ167.29,165.93,147.50,141.53,134.94,
134.36,131.88,131.63,131.20,130.14,129.56,118.05,111.31。
[0058]
c的总数目和理论上合成配体的c的总数目一致,都为15个c,其中δ=167.29pm(1c)与δ=165.93ppm(1c)是两个羧酸碳的特征吸收峰,118.05ppm(1c)处的单峰为氰基碳的的特征吸收峰,111.31ppm(1c)处的单峰为与氰基相连的苯环上碳的吸收峰,147.50ppm(1c)、141.53ppm(1c)、134.94ppm(1c)、134.36ppm(1c)、131.88ppm(1c)、131.63ppm(1c)、131.20ppm(1c)、130.14ppm(2c)、129.56ppm(2c)分别归属于苯环上另外11个碳的信号吸收峰。
[0059]
由以上可知,实施例中合成的最终产物为目标产物即2-氰基-4,4'-联苯二甲酸。
[0060]
实施例的作用与效果
[0061]
本实施例以联苯二甲酸二甲酯为原料,经过硝化反应、还原反应、重氮化反应、碘化反应、氰化反应及水解反应得到单氰基取代的多羧酸有机配体即2-氰基-4,4'-联苯二甲酸。本实施例提供的合成路线反应简单,普遍适用,通过氰基的引入,使得以后用该法合成金属有机骨架材料的种类得到丰富。
[0062]
另外,每一步反应收率都很高,水解反应的产率高达97%。
[0063]
此外,反应条件温和,所需温度为0℃—100℃,反应不需要无水无氧,操作十分方便,分离提纯只需层析分离纯化或抽滤洗涤即可,简单易行。
[0064]
此外,所采用的原料与溶剂价格便宜,易于得到,均为分析纯,且不需要任何后处理即可使用。
[0065]
另外,利用本实施例合成出来的2-氰基-4,4'-联苯二甲酸,可以进一步合成出氰基功能化的金属有机骨架材料。而该材料有着较大的比表面积和孔径,在气体吸附方面有着很大的应用,并且该材料的氰基官能团可以通过后合成修饰法进一步反应,形成其他功能化的金属有机骨架材料。
[0066]
上述实施方式为本发明的优选案例,并不用来限制本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1