一种获得高纯A晶型普伐他汀钠的制备方法与流程
本发明属于药物制备技术领域,具体涉及一种获得高纯a晶型普伐他汀钠的制备方法。
背景技术:
普伐他汀钠(pravastatinsodium),cas号为81131-70-6,是3-羟基-3-甲基戊二酰辅酶a还原酶(hmg-coa还原酶)抑制剂,结构式如式ⅰ。
通过抑制人体内胆固醇合成,普伐他汀钠可有效降低血清中总胆固醇、低密度脂蛋白和甘油三酯的含量,并升高高密度脂蛋白含量,最初用于治疗高血脂症和家族性高胆固醇。此外,普伐他汀钠还可以减缓动脉粥样硬化的发展,减少冠状动脉粥样硬化病变和临床心血管事件的发生,同时,也有研究显示,普伐他汀还能降低糖尿病和阿尔茨海默病(老年痴呆症)的发生率;抑制肝癌细胞的增殖;有效治疗溃疡性结肠炎,副作用少,半年复发率低;在治疗慢性心力衰竭、舒张性心力衰竭方方面可改善心功能,改善心室重塑等。
普伐他汀钠属于多晶型物,根据已报道的文献可知:普伐他汀钠的晶型具有a型、b型、c型、d型、e性、f型、g型、h型、h1型、i型、j性、k型、l型等多种晶型结构。相对比普伐他汀钠其他晶型,a晶型普伐他汀钠性质稳定,引湿性较缓慢,因此更有利于药品制剂的制备及药品制剂的保存,延长药品有效期。
目前,工业上生产普伐他汀钠的方法为两步发酵法。具体的方法包括:第一步,通过青霉属的一些微生物的培养获得前体化合物美伐他汀;第二步,通过微生物转化将美伐他汀羟化为普伐他汀钠。而现有技术从发酵液中纯化普伐他汀钠的方法主要采用有机溶媒对发酵液进行抽提,但是需要采用大量的有机溶媒,杂质含量高,纯化度低,给环境带来污染等问题,且无法判断为a晶型。中国专利cn1434702a公开了一种新的帕伐他丁形式,具体公开了帕伐他丁形式a及其制备方法,包括将任何固体形式的帕伐他丁钠溶解在质子溶剂中以形成溶液,用非质子传递溶剂将帕伐他丁钠溶液稀释和从帕伐他丁钠溶液中结晶出帕伐他丁钠形式a。已公开的a晶型普伐他汀钠制备工艺要么使用混合溶剂,溶剂回收难,生产成本高;要么毒性大,不适于药品生产;要么收率高,但纯度低。因此,如何从发酵液中得到具有高收率、高纯度的a晶型普伐他汀钠具有重要的意义。
技术实现要素:
本发明旨在提供一种获得高纯a晶型普伐他汀钠的制备方法,本发明能够定向制得a晶型普伐他汀钠,收率好,杂质含量少,安全性高,且制备方法工艺稳定,简单易控,便于工业化规模推广应用。
为了达到上述目的,本发明采用以下技术方案:
一种获得高纯a晶型普伐他汀钠的制备方法,包括以下步骤:
s1)含有普伐他汀的发酵培养液经板框过滤后收集滤液,调节ph小于5.0,用萃取剂萃取,得到萃取液;
s2)将步骤s1中的萃取液加热回流不少于1小时,降温至室温后用硅藻土层过滤,并用碳酸钠水溶液洗涤,洗涤后的溶液减压浓缩,降温至15℃以下结晶,过滤,减压干燥,得到普伐他汀内酯晶体;
s3)将步骤s2中的普伐他汀内酯晶体用甲基异丁酮在40~60℃下溶解,加入碳酸钠水溶液洗涤并于室温下搅拌不少于1小时,加入稀硫酸调ph为4.5~5.0,继续搅拌不少于1小时,分弃水层,加入二环已胺,降温结晶,过滤,得到普伐他汀胺盐湿固体;
s4)将步骤s3中的普伐他汀胺盐湿固体用甲基异丁酮在45~60℃下溶解,加入磷酸盐缓冲液并搅拌不少于1小时,分去水层,再加入纯化水洗涤,分弃水层,得到洗涤液;
s5)将步骤s4中的洗涤液加入氢氧化钠溶液并在室温、真空状态下搅拌不少于0.5小时,降温至10℃以下结晶,过滤,干燥,即得。
进一步地,所述步骤s1中的滤液与萃取剂的体积比为1:1。
进一步地,所述步骤s1中的萃取剂为乙酸乙酯、甲苯、乙酸正丁酯和乙酸异丁酯中的一种。
进一步地,所述步骤s2中碳酸钠水溶液的质量浓度为1%~2%,所述步骤s3中碳酸钠水溶液的质量浓度为10%~20%。
进一步地,所述步骤s2中的普伐他汀内酯晶体干燥失重(lod)≤1%。
进一步地,所述步骤s3中的普伐他汀内酯晶体与甲基异丁酮的质量体积比为1:10~20,g:ml;所述步骤s4中的普伐他汀胺盐湿固体与甲基异丁酮的质量体积比为1:10~20,g:ml。
进一步地,所述步骤s3中的普伐他汀内酯晶体与二环已胺的摩尔比为1:1。
进一步地,所述步骤s4中磷酸盐缓冲液的磷酸盐与步骤s2中的普伐他汀内酯晶体的摩尔比为1:1,磷酸盐缓冲液的ph为3.0-5.5。
进一步地,所述步骤s4中的普伐他汀胺盐湿固体与纯化水的质量体积比为1:8~10,g:ml。
进一步地,所述步骤s5中氢氧化钠溶液中的氢氧化钠与步骤s3普伐他汀内酯的摩尔比为1:1,氢氧化钠溶液的质量浓度为30%~40%。
本发明提供了任一上述制备方法得到的a晶型普伐他汀钠。
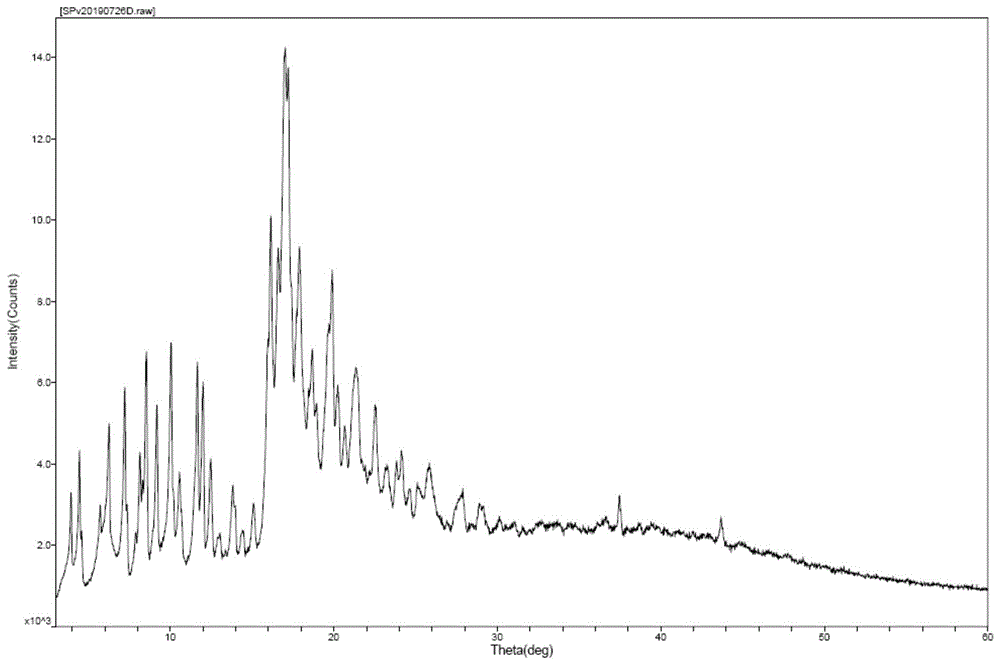
进一步地,所述a晶型普伐他汀钠的x-射线粉末衍射图谱在衍射角2θ°=3.9±0.2,4.5±0.2,6.2±0.2,7.2±0.2,8.6±0.2,9.2±0.2,10.0±0.2,11.6±0.2,12.0±0.2,17.0±0.2和20.0±0.2处有特征峰。
与现有技术相比,本发明具有以下有益效果:
(1)本发明利用含有普伐他汀的发酵培养液定向制得a晶型普伐他汀钠,由试验二可见,本发明制得的a晶型普伐他汀钠收率高,达到90%以上;由试验三可见,本发明制得的a晶型普伐他汀钠堆密度>0.30g/ml,晶体大小适宜,适用于制剂的生产;由试验四可见,本发明制得的a晶型普伐他汀钠最大单一杂质<0.1%,总杂质<0.6%,杂质含量少,安全性高。
(2)本发明a晶型普伐他汀钠的制备方法详细公开了每一步骤的具体操作和参数,工艺稳定,简单易控,便于工业化规模推广应用;且使用的溶剂毒性较小,对人体和环境影响较小。
附图说明
图1为实施例1a晶型普伐他汀钠的x-射线粉末衍射图谱。
具体实施方式
以下通过实施例形式的具体实施方式,对本发明的上述内容作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下实施例。
含有普伐他汀的发酵培养液为广东蓝宝制药有限公司提供,具体的方法可参照cn105821086a、一种规模化生产普伐他汀的工艺中的步骤s1。
实施例1、一种获得高纯a晶型普伐他汀钠的制备方法
包括以下步骤:
s1)含有普伐他汀的发酵培养液2300ml经板框过滤后收集滤液,滤液为2000ml,调节ph4.7,用2000ml乙酸乙酯萃取,得到萃取液;
s2)将步骤s1中的萃取液加热回流2h,降温至室温后用硅藻土层过滤,并用500ml,1%碳酸钠水溶液洗涤,洗涤后的溶液不高于50℃减压浓缩至300ml,降温至12℃结晶,过滤,减压干燥,得到普伐他汀内酯晶体22g;
s3)将步骤s2中的22g普伐他汀内酯晶体用220ml甲基异丁酮在60℃下溶解,加入碳酸钠水溶液洗涤并于室温下搅拌1.5h,加入稀硫酸调ph为4.5,继续搅拌1.5h,分弃水层,加入5.5ml二环已胺,降温至8℃结晶,过滤,得到普伐他汀胺盐湿固体30g;
s4)将步骤s3中的普伐他汀胺盐湿固体用450ml甲基异丁酮在55℃下溶解,加入1摩尔当量磷酸盐缓冲液(ph3.0)并搅拌,分去水层,再加入240ml纯化水洗涤,分弃水层,得到洗涤液;
s5)将步骤s4中的洗涤液加入5ml、40%氢氧化钠溶液并在室温、真空状态下搅拌,降温结晶,过滤,干燥(温度50~70℃、真空不低于0.09mpa的条件下),即得。
实施例2、一种获得高纯a晶型普伐他汀钠的制备方法
包括以下步骤:
s1)含有普伐他汀的发酵培养液2300ml经板框过滤后收集滤液,滤液为2000ml,调节ph4.5,用2000ml乙酸异丁酯萃取,得到萃取液;
s2)将步骤s1中的萃取液加热回流1.5h,降温至室温后用硅藻土层过滤,并用500ml,1.5%碳酸钠水溶液洗涤,洗涤后的溶液不高于50℃减压浓缩至300ml,降温至12℃结晶,过滤,减压干燥,得到普伐他汀内酯晶体23g;
s3)将步骤s2中的23g普伐他汀内酯晶体用345ml甲基异丁酮在50℃下溶解,加入碳酸钠水溶液洗涤并于室温下搅拌1.5h,加入稀硫酸调ph为4.5,继续搅拌1.5h,分弃水层,加入5.7ml二环已胺,降温至8℃结晶,过滤,得到普伐他汀胺盐湿固体33g;
s4)将步骤s3中的普伐他汀胺盐湿固体用594ml甲基异丁酮在50℃下溶解,加入1摩尔当量磷酸盐缓冲液(ph4.0)并搅拌,分去水层,再加入330ml纯化水洗涤,分弃水层,得到洗涤液;
s5)将步骤s4中的洗涤液加入5ml、35%氢氧化钠溶液并在室温、真空状态下搅拌,降温结晶,过滤,干燥(温度50~70℃、真空不低于0.09mpa的条件下),即得。
实施例3、一种获得高纯a晶型普伐他汀钠的制备方法
包括以下步骤:
s1)含有普伐他汀的发酵培养液2300ml经板框过滤后收集滤液,滤液为2000ml,调节ph4.5,用2000ml乙酸乙酯萃取,得到萃取液;
s2)将步骤s1中的萃取液加热回流2h,降温至室温后用硅藻土层过滤,并用500ml,1%碳酸钠水溶液洗涤,洗涤后的溶液不高于50℃减压浓缩至300ml,降温至12℃结晶,过滤,减压干燥,得到普伐他汀内酯晶体22g;
s3)将步骤s2中的22g普伐他汀内酯晶体用330ml甲基异丁酮在50℃下溶解,加入碳酸钠水溶液洗涤并于室温下搅拌2h,加入稀硫酸调ph为4.7,继续搅拌2h,分弃水层,加入5.5ml二环已胺,降温至8℃结晶,过滤,得到普伐他汀胺盐湿固体30g;
s4)将步骤s3中的普伐他汀胺盐湿固体用450ml甲基异丁酮在50℃下溶解,加入1摩尔当量磷酸盐缓冲液(ph5.0)并搅拌,分去水层,再加入270ml纯化水洗涤,分弃水层,得到洗涤液;
s5)将步骤s4中的洗涤液加入5ml、30%氢氧化钠溶液并在室温、真空状态下搅拌,降温结晶,过滤,干燥(温度50~70℃、真空不低于0.09mpa的条件下),即得。
实施例4、一种获得高纯a晶型普伐他汀钠的制备方法
包括以下步骤:
s1)含有普伐他汀的发酵培养液1100l经板框过滤后收集滤液,滤液为1000l,调节ph4.7,用1000l乙酸乙酯萃取,得到萃取液;
s2)将步骤s1中的萃取液加热回流2h,降温至室温后用硅藻土层过滤,并用200l,1%碳酸钠水溶液洗涤,洗涤后的溶液不高于50℃减压浓缩至150l,降温至12℃结晶,过滤,减压干燥,得到普伐他汀内酯晶体11kg;
s3)将步骤s2中的11kg普伐他汀内酯晶体用110l甲基异丁酮在60℃下溶解,加入碳酸钠水溶液洗涤并于室温下搅拌1.5h,加入稀硫酸调ph为4.5,继续搅拌1.5h,分弃水层,加入2.75l二环已胺,降温至8℃结晶,过滤,得到普伐他汀胺盐湿固体12.2kg;
s4)将步骤s3中的普伐他汀胺盐湿固体用183l甲基异丁酮在55℃下溶解,加入1摩尔当量磷酸盐缓冲液(ph5.5)并搅拌,分去水层,再加入549l纯化水洗涤,分弃水层,得到洗涤液;
s5)将步骤s4中的洗涤液加入2l、40%氢氧化钠溶液并在室温、真空状态下搅拌,降温结晶,过滤,干燥(温度50~70℃、真空不低于0.09mpa的条件下),即得。
对比例1、一种获得高纯a晶型普伐他汀钠的制备方法
与实施例1类似,区别在于,将步骤s3和步骤s4中的甲基异丁酮替换成丙酮,其他参数与实施例1相同。
对比例2、一种获得高纯a晶型普伐他汀钠的制备方法
与实施例1类似,区别在于,步骤s4中未添加磷酸盐缓冲液,其他参数与实施例1相同。
对比例3、一种获得高纯a晶型普伐他汀钠的制备方法
与实施例1类似,区别在于,步骤s3中未添加二环已胺,省略步骤s4,直接进行步骤s5,其他参数与实施例1相同。(由于步骤s3中未添加二环已胺,步骤s4无法操作)
试验一、a晶型普伐他汀钠测定
试验样品:实施例1~4和对比例1~3。
试验结果:如图1所示,为本发明的实施例1的xrd检测图谱,在2θ°=3.9±0.2,4.5±0.2,6.2±0.2,7.2±0.2,8.6±0.2,9.2±0.2,10.0±0.2,11.6±0.2,12.0±0.2,17.0±0.2和20.0±0.2处有特征吸收峰,确定为a晶型。实施例1~4为a晶型普伐他汀钠,对比例1为d晶型普伐他汀钠,对比例2~3为a晶型普伐他汀钠。
试验二、收率的计算
试验方法:称量a晶型普伐他汀钠的质量,为m;称量普伐他汀内酯晶体的质量,为m。
计算方法:
表1收率的计算结果
从表1可以看出,实施例1~4的a晶型普伐他汀钠收率较好。
试验三、堆密度的计算
试验方法:称量a晶型普伐他汀钠的质量,为m;将a晶型普伐他汀钠放置于容器中,容器的体积为v,进行检测。
计算方法:
表2堆密度的计算结果
从表2可以看出,实施例1~4的a晶型普伐他汀钠具有较好的堆密度,堆密度>0.30g/ml,使得a晶型普伐他汀钠在制剂生产过程中,具有较好的可压性和流动性。而对比例1的堆密度0.27<0.30g/ml,使得其在制剂生产过程中的可压性和流动性较差。
试验四、杂质检测
表3杂质检测的结果
从表3可以看出,实施例1~4的a晶型普伐他汀钠最大单一杂质<0.1%,总杂质<0.6%,杂质含量少安全性高,其中实施例1为本发明的最佳实施例,实施例4为本发明的中试试验,可见即使将本发明规模化生产也能达到较好的效果。对比例1杂质与实施例相当,但获得的是d晶型,且堆密度偏低。对比例2未加入磷酸盐缓冲液,普伐他汀胺盐不能有效转化为普伐他汀钠产品,导致收率低且杂质含量高。对比例3中普伐他汀不能形成普伐他汀胺盐和普伐他汀酸形式,也未经过纯化水洗涤过程,杂质不能有效去除,从而杂质含量高。
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
- 还没有人留言评论。精彩留言会获得点赞!