有机膦氢化合物的制备方法与流程
有机膦氢化合物的制备方法
【技术领域】
1.本发明涉及有机功能新材料化学品领域,首次披露了自有机膦卤(含有p-x键)化合物经由三氯硅烷还原生成相应有机膦氢(含有p-h键)产物的新方法。有机膦氢产物是已知的用途极为广泛的高附加值精细化学品。
背景技术:
2.含有活性膦-氢(p-h)键的有机膦氢化合物是应用广泛的高附加值新材料精细化学品,可以广泛用于制造膦酰型光引发剂,低烟无卤阻燃剂,电子化学品,农药和医药化学品等。工业上通常使用廉价易得的有机膦卤(p-x)前体的化学还原来制备,已知的还原剂例如固体形式的四氢铝锂或金属钠,属于成本高昂且极其湿气和空气敏感的材料,生产操作中安全风险和运营成本高企。但是,鉴于膦卤键还原的特殊性,一直未能发现更加安全有效同时成本低廉的还原剂体系,系业界当前一个重大共性技术瓶颈问题之一。
技术实现要素:
3.本项申请现已首次意外地发现,如下反应式(i)所示,形式为r1r2px的有机膦卤化合物在三氯氢硅(hsicl3)和适当反应条件conditions下还原能高效清洁且低成本地制备相应的有机膦氢产物r1r2ph。值得注意的是,三氯氢硅自身是极为廉价的大宗工业原材料,该工艺所涉及的原料和试剂通常都是液体,可以方便地通过管道输送和无人自动化的方式进行操作,是对有机膦氢化合物制备技术的一次革命。
[0004][0005]
其中x是卤素氯,溴,碘,氟;r1和r2分别独立的是卤素,或含有1-24个碳原子直链或支链的烷基,或含有4-24个碳原子的取代或未取代的(杂)芳基;三氯硅烷的使用量是1-100摩尔当量,优选的是1-50摩尔当量,更优选的是1-20摩尔当量。三氯硅烷在本反应中可以既做还原剂又充作溶剂。
[0006]
conditions是溶剂,温度,压力,和/或添加剂中任意之一或任意二者或二者以上的联合应用。
[0007]
本工艺所涉及的反应“溶剂”选自含有1-24个碳的取代或非取代的芳香烃,直链或支链的脂肪烃,酰胺,醚,酯,酮,腈,羧酸,水,胺,离子液,超临界二氧化碳,或上述任意二者或二者以上类型溶剂组成的混合溶剂;优先的溶剂是三氯硅烷,二氯甲烷,二氯乙烷,氯仿,四氯化碳,苯,甲苯,二甲苯,三甲苯,四甲苯,乙腈,乙苯,二乙苯,氯苯,二氯苯,苯甲醚,硝基苯,庚烷,己烷,石油醚,二氧六环,四氢呋喃,甲基叔丁基醚,乙二醇二甲醚,双缩乙二醇二甲醚,三缩乙二醇二甲醚,丙二醇甲醚醋酸酯,三乙胺,三丁胺,二甲基异丙胺,吡啶,n,n-四甲基乙二胺,n-烷基吗啉,n-烷基吡咯,n,n-二甲基甲酰胺,甲酰吗啉,n,n-二乙基甲酰胺,n-甲基吡咯烷酮,或上述任意二者或二者以上溶剂组成的混合溶剂。
[0008]
本工艺所涉及的反应“温度”选自-70摄氏度至200摄氏度之间,优先的是选自-30
摄氏度至180摄氏度之间,更优先的是选自-20摄氏度至150摄氏度之间。
[0009]
本工艺所涉及的反应“压力”选自0.001至200个标准大气压之间,优先的是选自0.1至100个标准大气压之间,更优先的是选自0.1至20个标准大气压之间。
[0010]
本工艺所涉及的反应“添加剂”涵盖反应促进剂,增效剂,催化剂,和/或功能助剂,其是路易斯酸或路易斯碱型(lewis acids/bases)单质或化合物,优选的是有机(叔)胺,碱金属,碱土金属,主族金属,或过渡金属的氟化物,氯化物,溴化物,碘化物,氧化物,氢氧化物,硫化物,烷氧化物,烷基或芳基金属化合物;或碱金属,碱土金属,主族金属,或过渡金属的碳酸盐,碳酸氢盐,亚硫酸盐,硫酸氢盐,磺酸盐,或羧酸盐;或碱金属,碱土金属,或过渡金属的一元或多元有机磷,有机胺,羟基,酮羰基,酯羰基,或羧酸配体的配合物;或四烷基卤化铵,四烷基氢氧化铵,或离子液体(ionic liquids);或无机质子酸,有机羧酸,有机磺酸,杂多酸,分子筛,沸石,硅藻土,蒙拓土,高岭土;或硼,硅,磷元素的氟化物,氯化物,溴化物,碘化物,氧化物,氢氧化物,硫化物,烷氧化物;或满足上述定义“添加剂”中任意二者或二者以上的混合物或其联合使用。以反应原材料摩尔当量为基准,所述“添加剂”的使用量可以是催化量,等当量,或过当量。
[0011]
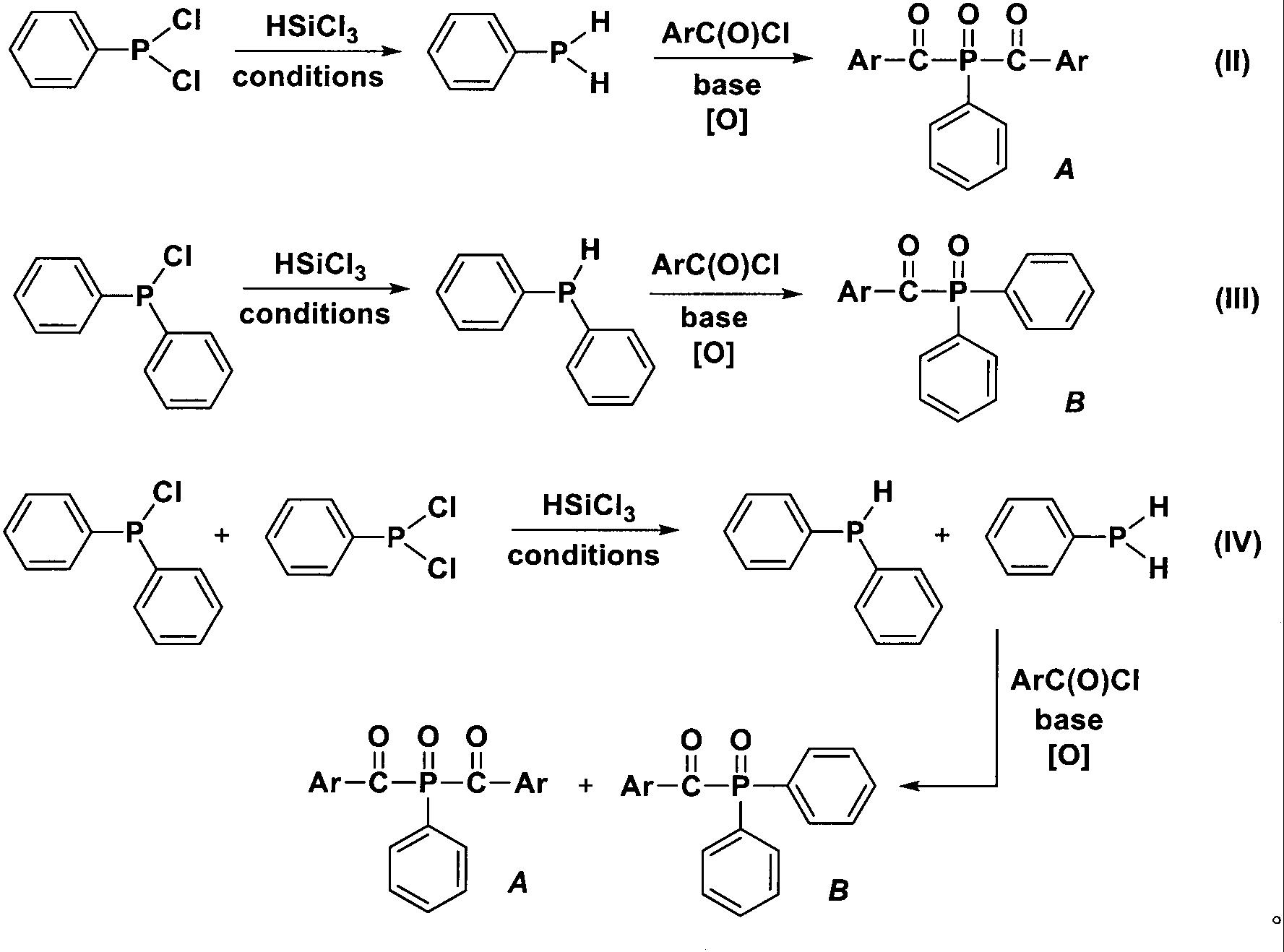
鉴于有机膦氢化合物和酰氯在碱的促进下缩合生成酰基膦中间体(专利cn1198831),进而氧化成为酰基膦氧(acyl phosphine oxide)产物是文献已知的技术,本发明批露的工艺可直接用于制造双酰基膦氧化合物光引发剂a(反应式ii),单酰基膦氧化合物光引发剂b(反应式iii),以及同时“一锅法”联产上述化合物a和b(反应式iv)。这里ar是芳基或叔丁基,优选的ar是2,4,6-三甲基苯甲酰基;base是无机碱或有机叔胺;【o】是氧化剂,优选的氧化剂是h2o2或o2。
[0012][0013]
在实施例中我们将进一步说明。
【具体实施方式】
[0014]
实施例一:苯基膦的合成
[0015][0016]
在氮气保护下,将17.9克一苯基二氯化磷和300毫升乙腈加入到1升三口瓶中,室温下加入32.3克二异丙基乙基胺,后冰浴下滴加33.9克三氯硅烷于100毫升的乙腈溶液,加毕由0℃缓缓升至80℃搅拌反应过夜。随后反应体系降温至0℃并缓慢加入300ml预先仔细脱氧处理的饱和氢氧化钾溶液,脱氧乙酸乙酯萃取,有机相用脱氧饱和食盐水洗涤、mgso4干燥后浓缩得到淡黄色粗品,随后粗品进行减压蒸馏得目标产物一苯基膦9.13克,产率83%(核磁共振
31
p-nmr特征吸收峰在-122.7ppm)。叔胺助剂即二异丙基乙基胺可以在减压蒸馏过程中回收套用。
[0017]
实施例二:苯基膦的合成
[0018][0019]
在氮气保护下,将35.8克一苯基二氯化磷和500毫升四氢呋喃加入到2升三口瓶中,室温下加入64.6克二异丙基乙基胺,后冰浴下滴加67.8克三氯硅烷于200毫升的四氢呋喃溶液,加毕由0℃缓缓升温至回流搅拌反应过夜。随后反应体系加入500毫升预先脱氧处理的饱和氢氧化钾溶液,脱氧乙酸乙酯萃取,有机相用脱氧饱和食盐水洗涤、mgso4干燥后浓缩得到粗品。随后粗品进行减压蒸馏得目标产物一苯基膦19.1克,产率87%。
[0020]
实施例三:二苯基膦的合成
[0021][0022]
在氮气保护下,将22克二苯基氯化磷和300毫升四氢呋喃加入到1升三口瓶中,室温下加入19.4克二异丙基乙基胺,后冰浴下滴加20.3克三氯硅烷于100毫升的四氢呋喃溶液,加毕由0℃缓缓升温至回流搅拌反应过夜。随后反应体系加入300ml预先仔细脱氧处理的饱和koh溶液,脱氧乙酸乙酯萃取,有机相用脱氧饱和食盐水洗涤、mgso4干燥后浓缩得到粗品。随后粗品进行减压蒸馏得目标产物二苯基膦16.9克,产率91%(核磁共振
31
p-nmr特征吸收峰在-40.4ppm)。
[0023]
实施例四:双膦酰氧光引发剂819合成(ar=2,4,6-三甲基苯甲酰基)
[0024][0025]
参照实施例一的步骤,将2.5克新鲜制备和蒸馏纯化的苯基膦直接接收在120ml二甲苯冷阱溶液中,在室温和氮气下向其依次分批加入4.5克的叔丁醇钠和9.1克的2,4,6-三
甲基苯甲酰氯,搅拌反应2小时,滴加浓硫酸调节体系为酸性,进而慢慢滴加7ml的30%双氧水。体系用20ml水稀释,有机相分离后分别用10%碳酸氢钠溶液和水洗涤两次,有机相用无水硫酸钠干燥,过滤,减压浓缩,得到亮黄色固体用己烷打浆,得到粉末目标产品8.8克。
[0026]
实施例五:单膦酰氧光引发剂tpo合成(ar=2,4,6-三甲基苯甲酰基)
[0027][0028]
参照实施例三的步骤,将3.6克新鲜制备和蒸馏纯化的二苯基膦直接接收在100ml二甲苯冷阱溶液中,在室温和氮气下向其依次分批加入2.0克的叔丁醇钠和3.7克的2,4,6-三甲基苯甲酰氯,搅拌反应2小时,滴加浓硫酸调节体系为酸性,进而慢慢滴加4.5ml的30%双氧水。体系用30ml水稀释,有机相分离后分别用10%碳酸氢钠溶液和水洗涤两次,有机相用无水硫酸钠干燥,过滤,减压浓缩,得到淡黄色固体用乙醇结晶得到目标产品6.2克。
[0029]
实施例六:tpo和819的“一锅法”联产(ar=2,4,6-三甲基苯甲酰基)
[0030][0031]
参照上述实施例的步骤条件,将20.2克的苯基二氯化膦和25.0克二苯基氯化膦的混合物和73克三氯硅烷“一锅煮”还原得到的苯基膦和二苯基膦混合物直接减压蒸馏到800ml二甲苯冷阱溶液中,在室温和氮气下向其依次分批加入35克的叔丁醇钠和65克的2,4,6-三甲基苯甲酰氯,搅拌反应4小时,滴加浓硫酸调节体系为酸性,进而慢慢滴加140ml的30%双氧水。体系用约半体积水稀释,有机相分离后分别用10%碳酸氢钠溶液和水洗涤两次,有机相用无水硫酸钠干燥,过滤,减压浓缩,得到的粘稠黄色液体用己烷打浆,得到黄色固体粉末,干燥后得到目标产品819和tpo的混合物71.2克,二者含量比例是46/54。
[0032]
需要强调的是,上述实施例仅仅为示例性而非限定性说明,基于本项申请披露,任何从业技术人员所通常可能采用的反应条件或参数等调整或变动均不会偏离本发明的要旨,本专利的保护范围应以相关的权利书记载条目为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1