一种聚酯酰亚胺聚合物的制备方法
1.本发明属于聚合物高分子材料及其合成技术领域,具体涉及一种聚酯酰亚胺聚合物的制备方法。
背景技术:
2.聚酯酰亚胺是一类以酯键和酰亚胺键为基础构建单元的新型聚合物。独特的分子结构使其有利于协调统筹传统聚酯和聚酰亚胺两者的优势,赋予其优异的热稳定性、绝缘性、电磁屏蔽性、耐溶剂性以及出色的机械性能。优异的性能使聚酯酰亚胺在电子、航空航天、电路印刷等领域具有广阔的应用前景。目前,合成聚酯酰亚胺的方法主要可分为两类:1)第一类是先制备含酯基的双胺或双酸酐单体,然后再依照聚酰亚胺制备思路进行酰亚胺化聚合反应及后续加工。由于该方法的氨基在反应过程中会不可避免地破坏单体中预先设计好的酯键,从而扰乱最终分子结构。因此,该方法难以精确控制产物分子结构,得到的产物为含有多种异构体的混合物。2)第二类则是预先将制备含有酰亚胺环的单体,以单体上预留的羟基或羧基为活性基团通过常规聚酯合成,得到结构精确的产物。比如,中国发明专利cn105085912 b,公开了一种透明聚酯酰亚胺树脂的制备方法;具体为:(1)将二元伯胺单体加到氮气或低氧气体保护的极性非质子溶剂中,搅拌使其完全溶解后加入对苯二甲酸二(3,4-二甲酸酐)苯酯,搅拌反应12小时~48小时,得透明粘稠的聚酯酰胺酸溶液;(2)采用热亚胺化法或化学亚胺化法将聚酯酰胺酸溶液制备成聚酯酰亚胺溶液,干燥,制得透明聚酯酰亚胺树脂。该发明所制备的聚酯酰亚胺在玻璃化转变温度上比原有的聚醚酰亚胺有所提高。然而,当前合成方法都面临着同样的问题,即合成过程需采用大量溶剂或特定催化剂,这不仅对生产设备提出了更高的要求和增加能耗,而且会大幅提高了企业生产成本和环评压力。此外,采用预先合成特定单体的工艺和后处理工序也使得合成过程过于冗长繁琐,不利于大范围推广和使用,从而成为制约聚酯酰亚胺研究和应用的瓶颈。
技术实现要素:
3.本发明的目的是提供一种聚酯酰亚胺聚合物的制备方法。本发明的制备方法操作简单、反应条件温和、无需贵金属或催化剂参与、无溶剂参与、成本低、对设备要求低;制备得到的聚合物中的酰亚胺键与酯键以交替形式排列、结构均一、分子量可控、分子量分布窄。
4.本发明的聚酯酰亚胺聚合物的制备方法;第一步先在设定温度和压力的低氧或惰性气体氛围下将偏苯三酸酐与乙醇胺进行预混合,进行酰胺化反应;第二步升高温度,在一定温度和压力下同时进行熔融聚酯反应和酰亚胺化反应获得聚酯酰亚胺产品。需要说明的是中国发明cn201610739154.1公开了一种含羧基高折射率超支化聚酯酰胺的制备方法,其是在催化剂的作用下制备结构为超支化的聚酯酰胺目标产物,而本发明则是利用酰亚胺化和酯化的自身活性差异的特点制备线性结构的聚酯酰亚胺目标产物,两者从反应机理到目标产品分子结构均为不同。本技术打破了常规思维对此类反应进程和机理的理解,并从实
际实验角度论证了该技术的可行性和正确性,而在未经实验论证下所涉及本领域的技术人员不易想到本技术的巧妙性和可行性。
5.具体而言,本发明的目的是通过以下技术方案来实现的:
6.第一方面,本发明涉及一种聚酯酰亚胺聚合物的制备方法,所述方法包括如下步骤:
7.s1、先将偏苯三酸酐与乙醇胺在低氧或惰性气体氛围下搅拌预混合,进行酰胺化反应;
8.s2、升高温度,在预设温度和压力下同时进行熔融聚酯反应和酰亚胺化反应,反应结束后即得聚酯酰亚胺聚合物。
9.作为本发明的一个实施方案,步骤s1中,先将乙醇胺投入聚酯合成釜中,随后分批加入偏苯三酸酐。
10.作为本发明的一个实施方案,步骤s1中,所述低氧或惰性气体为空气、氮气、氩气、氦气中的一种或几种的组合。
11.作为本发明的一个实施方案,步骤s1中,偏苯三酸酐与乙醇胺的摩尔比为0.5~2:1。
12.作为本发明的一个实施方案,步骤s1中,料液温度控制在50℃~200℃之间。
13.作为本发明的一个实施方案,步骤s1中,酰胺化反应至物料状态呈固液混合的浆状或固体状。
14.作为本发明的一个实施方案,步骤s1中,搅拌时间为5~240min。
15.作为本发明的一个实施方案,步骤s1中,反应压力为0.05~20mpa。
16.作为本发明的一个实施方案,步骤s2中,升高温度至220℃~280℃。
17.作为本发明的一个实施方案,步骤s2中,反应压力为0~50mpa。作为一个优选方案,反应压力为0~20mpa。作为另一个优选方案,采用两步阶梯式降压反应。更优选先在常压下反应10~120min,随后抽真空至10~100pa,继续反应。
18.作为本发明的一个实施方案,步骤s2中,根据出水量判断反应进程;以占总投料质量的百分比为计,出水量为2%~15%时,反应完成。
19.作为本发明的一个实施方案,步骤s2中,反应时间为10~180min。
20.第二方面,本发明涉及一种聚酯酰亚胺聚合物,所述聚酯酰亚胺聚合物的结构式如式(ⅰ)所示:
21.式中,n=1~100。
22.所述聚酯酰亚胺聚合物是根据上述制备方法合成得到的。
23.与以往报道的聚酯酰亚胺合成技术相比,本发明具有如下有益效果:
24.1)本发明提出的合成技术采用易得的商品化原料偏苯三酸酐和乙醇胺为起始原料,经预混合和熔融聚合两步得到最终产物,全程操作简单、无需进行分离提纯、反应条件温和、无溶剂参与、无需贵金属或催化剂参与、成本低、对设备要求低、易于推广;
25.2)本发明合成得到的聚合物分子中酰亚胺键与酯键以交替形式排列、结构规整、分子量可控、分子量分布窄。
附图说明
26.通过阅读参照以下附图对非限制性实施例所作的详细描述,本发明的其它特征、目的和优点将会变得更明显:
27.图1是实施例1产物的核磁氢谱图;
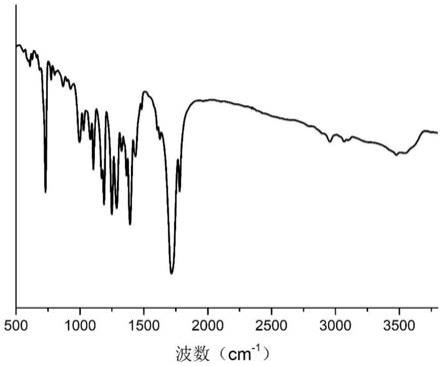
28.图2是实施例1产物的红外光谱图;
29.图3是实施例1产物的maldi-tof图;
30.图4是实施例1产物的dsc升温曲线图;
31.图5是实施例1产物的tga曲线图。
具体实施方式
32.下面结合实施例对本发明进行详细说明。以下实施例将有助于本领域的技术人员进一步理解本发明,但不以任何形式限制本发明。应当指出的是,对本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干调整和改进。这些都属于本发明的保护范围。并且,实施本发明的过程、条件、试剂、实验方法等,除以下专门提及的内容之外,均为本领域的普遍知识和公知常识。
33.本发明提供一种新型聚酯酰亚胺聚合物的制备方法,先将偏苯三酸酐与乙醇胺进行预混合,进行部分酰胺化反应,随后采用熔融聚酯反应和酰亚胺化反应一锅制备聚酯酰亚胺聚合物。其合成步骤包括以下两步:
34.(a)在低氧或惰性气氛下,开启搅拌,先将乙醇胺投入带机械搅拌的三口烧瓶中,随后分批加入偏苯三酸酐,搅拌至物料成特定状态。
35.(b)在上一步反应达到所需状态后,开启升温模式,料液在高温下同时进行熔融聚酯反应和酰亚胺化反应,根据出水量(以占总投料质量的百分比为计)判断反应进程。待反应完成后可得到最终聚酯酰亚胺聚合物。
36.再通过下面的实施例对本发明的技术予以进一步的说明。
37.实施例1
38.第一步:将19.06g乙醇胺加入到带有机械搅拌的三口烧瓶中,氮气置换三次;开启搅拌,在氮气氛围下,加入60.00g偏苯三酸酐;开启加热套,确保反应体系温度为50℃,在此温度下搅拌120min。
39.第二步:将加热套升至220℃,常压下反应30min,同时收集副产物水。待反应完成后得到最终聚酯酰亚胺聚合物71.15g,产率约为90%,副产物水7.90g,出水量为10%。
40.核磁氢谱(图1)显示,3.60ppm和3.66ppm两处的峰为末端亚甲基的质子特征峰,4.00ppm和4.50ppm附近两处峰为主链上亚甲基的质子特征峰;红外图谱(图2)显示,713cm-1
、1078cm-1
、1365cm-1
波数处的峰为酰亚胺环的特征峰,1720cm-1
附近的峰为酯基和酰亚胺上羰基的伸缩振动特征峰。两种测试结果表明目标聚合物是由酰亚胺和酯键交替连接构成的。maldi-tof测试结果显示,目标聚合物的重复单元分子量为217,与偏苯三酸酐和乙醇胺脱去两分子水缩合得到的重复单元分子量相吻合,且根据测试数据分布可计算其数均分子
量、质均分子量和分子量分布。上述所有结果证明了式(ⅰ)所示结构的正确性。
41.dsc测定聚合物tg为105℃(图4),tga测定其分解温度为440℃(图5),maldi-tof测定其数均分子量为1500(图3),分子量分布为1.13。
42.实施例2
43.本实施例中的偏苯酸酐与乙醇胺的摩尔用量比为2:1,其他条件与实施例1相同,产率为98%,出水量约为2%,dsc测定聚合物tg为90℃,tga测定其分解温度为400℃,maldi-tof测定其数均分子量为500,分子量分布为1.10。
44.实施例3
45.本实施例中的偏苯酸酐与乙醇胺的摩尔比为1:2,其他条件与实施例1相同,产率为67%,出水量和未反应的乙醇胺共约为33%,dsc测定聚合物tg为89℃,tga测定其分解温度为380℃,maldi-tof测定其数均分子量为700,分子量分布为1.14。
46.实施例4
47.本实施例中第一步的温度设置为200℃,搅拌5min,当物料状态转变成固态时开始进行第二步,其他条件与实施例1相同,产率为90%,出水量为10%,dsc测定聚合物tg为105℃,tga测定其分解温度为440℃,maldi-tof测定其数均分子量为1500,分子量分布为1.17。
48.实施例5
49.本实施例中第二步的温度设置为280℃,其他条件与实施例1相同,产率为87%,出水量为13%,dsc测定聚合物tg为120℃,tga测定其分解温度为440℃,maldi-tof测定其数均分子量为3500,分子量分布为1.22。
50.实施例6
51.本实施例中第二步的反应时间为180min,最终出水量约为13%,其他条件与实施例1相同,产率为87%,出水量为13%,dsc测定聚合物tg为143℃,tga测定其分解温度为440℃,maldi-tof测定其数均分子量为3000,分子量分布为1.25。
52.实施例7
53.本实施例中第二步中,先在常压下反应30min,随后抽真空至20pa在该压力下继续反应1h,其他条件与实施例1相同,产率为86%,出水量为14%,dsc测定聚合物tg为160℃,tga测定其分解温度为440℃,maldi-tof测定其数均分子量为22000,分子量分布为1.3。
54.以上对本发明的具体实施例进行了描述。需要理解的是,本发明并不局限于上述特定实施方式,本领域技术人员可以在权利要求的范围内做出各种变形或修改,这并不影响本发明的实质内容。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1