自修复新型聚酯及其制备方法与流程
1.本发明涉及高分子材料技术领域,具体涉及自修复新型聚酯及其制备方法。
背景技术:
2.2-甲基-1,3-丙二醇(mpo)是一种低毒低粘度、无色透明的液体二元醇,mpo的甲基侧基结构和溶剂相容性较其他二元醇更好,分子结构独特,性能优良,可选择多种二元酸制备聚酯,发展前景广阔。
3.目前,mpo主要作为第三单体与其他二元醇和二元酸制备聚酯,mpo的加入量较少,采用mpo作为主要单体与二元酸直接反应制备的聚酯未见公开报道。
技术实现要素:
4.为了克服上述问题,本发明人进行了锐意研究,研究出一种自修复新型聚酯及其制备方法,该自修复新型聚酯由2-甲基-1,3-丙二醇与二甲酸单体制备得到,该自修复新型聚酯的mw/mn为1.2~1.6,特性粘度大于0.7dl/g,该自修复新型聚酯为非晶化合物,具有形状可恢复性(或称为自修复性),良好的可纺性,由该自修复新型聚酯所制备的新型聚酯纤维具有高湿热收缩率,具有良好的应用前景,该自修复新型聚酯制备方法简单、原料易得,可用于大规模工业化生产,从而完成本发明。
5.本发明的目的一方面在于提供一种自修复新型聚酯,所述新型聚酯的mn为20000~30000,mw为30000~40000,mw/mn为1.2~1.6,
6.所述自修复新型聚酯的特性粘度大于0.7dl/g。
7.所述自修复新型聚酯由2-甲基-1,3-丙二醇和二甲酸单体聚合制得,所述2-甲基-1,3-丙二醇与二甲酸单体的摩尔比为(1.2~1.8):1。
8.所述自修复新型聚酯由包括以下步骤的方法制备:
9.步骤1、酯化反应:将2-甲基-1,3-丙二醇与二甲酸单体加入到反应容器中,进行酯化反应,得到酯化产物;
10.步骤2、缩聚反应:将酯化产物进行缩聚,得到自修复新型聚酯。
11.本发明的另一方面在于提供一种自修复新型聚酯的制备方法,所述方法包括以下步骤:
12.步骤1、酯化反应:将2-甲基-1,3-丙二醇与二甲酸单体加入到反应容器中,进行酯化反应,得到酯化产物;
13.步骤2、缩聚反应:将酯化产物进行缩聚,得到自修复新型聚酯。
14.步骤1中,2-甲基-1,3-丙二醇与二甲酸单体采用直接酯化法,得到酯化产物,
15.所述二甲酸单体选自对苯二甲酸、或对苯二甲酸与间苯二甲酸的混合酸;
16.所述酯化反应的温度为200~260℃,排水期间控制反应容器内压力为200~330kpa。
17.在酯化反应结束后,缩聚反应前,加入缩聚催化剂,优选地,在酯化反应结束后,缩
聚反应前,将反应体系抽真空10-60min,再加入缩聚催化剂。
18.所述缩聚催化剂为钛系催化剂,优选选自钛酸四丁酯、钛酸四乙酯、钛酸四正丙酯、钛酸四异丙酯或钛酸四异辛酯中的一种或几种。
19.所述缩聚催化剂的加入质量为二甲酸单体质量的0.1~1%。
20.步骤2中,缩聚反应温度为210~280℃,缩聚反应真空度为10~100pa。
21.在缩聚反应前还加入助剂,所述助剂包括热稳定剂、抗氧化剂中的一种或两种,
22.所述热稳定剂的加入量为二甲酸单体质量的0.1~0.5%,
23.所述热稳定剂为磷酸三甲酯、亚磷酸三苯酯、磷酸三苯酯、亚磷酸三甲酯中的一种或几种。
24.本发明所具有的有益效果为:
25.(1)本发明提供了一种自修复新型聚酯,该自修复新型聚酯由2-甲基-1,3-丙二醇与二甲酸单体制备得到,所得自修复新型聚酯的mw/mn为1.2~1.6;
26.(2)本发明提供该自修复新型聚酯的制备方法,探索出制备该自修复新型聚酯适用的缩聚催化剂及其用量,以及缩聚催化剂的加入时间与加入方式,使得催化效果较好,且缩聚反应稳定且可控;
27.(3)本发明以2-甲基-1,3-丙二醇为醇单体制备自修复新型聚酯,填补了以2-甲基-1,3-丙二醇为主要的醇单体制备自修复新型聚酯的空白,拓宽了其应用,并且,所得自修复新型聚酯具有良好的形状可恢复性,经过弯折的自修复新型聚酯在温度55℃以上具有极高的形状恢复率,能达到99.3%以上;
28.(4)本发明的自修复新型聚酯具有良好的可纺性,所制备的新型聚酯纤维湿热收缩率高,具有良好的应用前景;
29.(5)本发明的自修复新型聚酯的制备方法简单、易于实现,可大规模工业化生产。
附图说明
30.图1示出本发明实验例1所得红外谱图;
31.图2示出本发明实验例3所得gpc色谱图和分子量分布视图;
32.图3示出本发明实验例4中实施例1-7所得自修复新型聚酯的一次升温dsc曲线;
33.图4示出本发明实验例4中实施例1-2所得自修复新型聚酯在100℃退火处理的2h、4h和6h的dsc升温曲线;
34.图5示出本发明实验例4中实施例1-7所得自修复新型聚酯的tg曲线;
35.图6示出图5中的局部放大图;
36.图7示出实验例5中实施例10所得自修复新型聚酯在225℃、230℃和235℃下的剪切速率与剪切粘度关系;
37.图8示出实验例5中实施例13所得自修复新型聚酯在225℃、230℃和235℃下的剪切速率与剪切粘度关系;
38.图9示出本发明实验例6中自修复新型聚酯样条变形前和变形后的图片。
具体实施方式
39.下面通过附图和优选实施方式对本发明进一步详细说明。通过这些说明,本发明
的特点和优点将变得更为清楚明确。
40.根据本发明,提供一种自修复新型聚酯,该自修复新型聚酯由2-甲基-1,3-丙二醇(mpo)与二甲酸单体制备得到,优选地,使2-甲基-1,3-丙二醇与二甲酸单体进行酯化反应并得到酯化产物,使酯化产物进行缩聚反应制得自修复新型聚酯。
41.根据本发明,所述二甲酸单体选自对苯二甲酸或对苯二甲酸与间苯二甲酸的混合酸,优选为对苯二甲酸(pta)。
42.根据本发明,由mpo和pta制备得到自修复新型聚酯mptt。
43.根据本发明一种优选的实施方式,所述酯化产物具有如式(i)所示结构:
[0044][0045]
根据本发明,所述自修复新型聚酯具有如下式(ii)所示的结构:
[0046][0047]
根据本发明的优选实施方式,以2-甲基-1,3-丙二醇为二醇单体与对苯二甲酸直接酯化、缩聚反应,不添加其他醇单体,得到自修复新型聚酯。发明人理论推测:同ptt分子结构相比,mptt上侧链甲基的存在破坏了分子结构的完整性,大分子链结构不对称性导致该聚酯不易结晶,当温度达到能够使分子链链段开始运动的温度,由于非晶结构对分子链的束缚较小,分子链更易运动,同时甲基的存在加大了空间位阻效应,内排斥力增加,分子链趋向于向排斥力较小的方向移动,使得mptt材料获得自修复能力。当然,本发明的新型聚酯具有良好自修复能力的机制完全不受上述理论所束缚。
[0048]
该自修复新型聚酯的特性粘度大于0.7dl/g,优选能达到0.8dl/g以上,甚至可达到0.86dl/g,具备较好的纤维加工性能,可用于制备新型聚酯纤维。
[0049]
根据本发明,提供一种自修复新型聚酯的制备方法,优选为制备本发明第一方面所述的自修复新型聚酯的方法,该方法包括:
[0050]
步骤1、酯化反应:将mpo与二甲酸单体加入到反应容器中,进行酯化反应,得到酯化产物。
[0051]
根据本发明,步骤1中,mpo与二甲酸单体采用直接酯化法进行酯化反应,得到酯化产物,优选地,采用间歇的直接酯化法,灵活方便,适用于开发功能性聚酯。
[0052]
本发明人发现,在酯化反应过程中,醇酸投料比即mpo与二甲酸单体的投料摩尔比对酯化反应具有较大影响,醇酸投料摩尔比过大会导致两分子缩合形成醚化合物,摩尔比过小则会导致酯化不完全,酯化产物中羧基含量过高,酯化率低,达不到工艺规定的要求。实际生产中,需根据浆料(mpo与二甲酸单体液固混合物)的粘度、浆料配置设备结构、浆料输送管道结构和输送工艺等情况及其他可能的要求(如酯化工艺塔二醇是否回流)对最佳醇酸投料摩尔比进行调整。
[0053]
根据本发明,mpo与二甲酸单体的摩尔比为(1.2~1.8):1,优选为(1.2~1.6):1,更优选为(1.2~1.3):1。
[0054]
本发明人发现,酯化温度对该酯化反应具有重要影响,适当提高酯化反应温度有利于提高酯化反应速度,但同时也会提高副反应速度,酯化反应温度低会造成酯化反应速度慢、酯化反应不完全、端羧基含量过高、酯化率不达标、缩聚无法达到足够的分子量等现象,从而导致所得新型聚酯mptt产品质量降低,性能较差。
[0055]
根据本发明,酯化反应的温度为200~260℃,优选为205~250℃,更优选为240~248℃。
[0056]
根据本发明一种优选地实施方式,在2l的聚合反应釜中,酯化反应的温度为205~245℃,优选为240~246℃;
[0057]
在30l的聚合反应釜中,酯化反应的温度为220~250℃,优选为245~247℃。
[0058]
本发明人发现,酯化反应的压力与温度相互影响,由于mpo的沸点低于酯化反应的温度,酯化反应压力低时,会有部分mpo与体系生成的水形成共沸物在分馏柱中反复冷凝与汽化,不仅会消耗更多的能量,也会降低反应速率;酯化分馏柱顶温比较难以控制,酯化水中mpo的含量也较高,酯化反应的最理想状态是mpo尽可能多的存在于液相体系中,有利于mpo与对苯二甲酸尽快反应,形成的共沸物越少越有利于酯化反应的快速进行;
[0059]
酯化反应压力高时形成的共沸物少,易于控制柱顶温度,且排出的酯化水中醇的含量低;由于酯化压力由酯化水和醇的共沸物产生,酯化反应压力过高则酯化水用于维持反应体系的压力,不利于酯化水的排出,反而延缓了酯化反应进行,尽管增加mpo的浓度有利于酯化物的生成,但也增加了醚化合物的生成量,因此,压力的选择既要满足水的排除,又能保证酯化反应需要的物质的量(摩尔)比和减少醚化合物的生成量。
[0060]
根据本发明,酯化反应的初始压力为80~120kpa,优选为90~110kpa,更优选为100kpa。
[0061]
根据本发明,排水期间控制反应容器内压力为200~330kpa,优选为200~300kpa,在上述压力下,有利于排除酯化反应生产的水,且对酯化体系的温度影响最小,即压力波动较大时,釜内温也会产生较大波动,不利于反应的稳定进行和工艺的控制。
[0062]
本发明中,酯化反应时间包括正压反应时间和常压反应时间,酯化反应时间受搅拌速度、酯化反应温度、酯化反应压力和排水速率等因素的影响。酯化反应从开始加压到压力降为常压的时间称之为正压酯化反应时间,由反应釜压力和柱顶温度决定。一般情况下,当反应釜温度不低于230℃,酯化反应压力不再上升(或上升很缓慢),说明酯化反应生成的水量已不足以产生270kpa的釜内压力,此时,就需要将酯化反应压力主动逐渐降低,直至成为常压,降压过程约需30-50min,降压过程需要确保不会导致反应釜温度降低和产生较大的波动。酯化反应压力降为1个大气压至常压酯化反应结束的时间称之为常压酯化反应时
间。待酯化水出水口不再出水,且柱顶温度低于60℃时即判定常压酯化反应结束。
[0063]
根据本发明,酯化反应的正压反应时间为60~250min,常压反应时间为30~200min。
[0064]
根据本发明一种优选的实施方式,在2l的聚合反应釜中,酯化反应的正压反应时间为60~150min,常压反应时间为30~70min。
[0065]
根据本发明一种优选的实施方式,在2l的聚合反应釜中,酯化反应的正压反应时间为120~250min,常压反应时间为40~200min。
[0066]
根据本发明一种优选的实施方式,在2l聚合反应釜中,酯化温度为215~240℃,酯化压力为200~270kpa时,酯化反应的正压反应时间为70~140min,常压反应时间为40~60min。
[0067]
根据本发明一种优选的实施方式,在30l聚合反应釜中,酯化温度为230~247℃,酯化压力为200~300kpa时,酯化反应的正压反应时间为140~240min,优选150~180min,常压反应时间为40~180min,优选为150~180min。
[0068]
本发明中,酯化反应出水需达到理论出水量的93%以上,增大酯化反应压力和延长加压时间不利于排水,无助于加快酯化反应的进行。常压反应时间过短可能导致酯化反应不完全。需要综合考虑酯化反应温度、压力、出水速率及出水量来控制酯化反应。
[0069]
本发明中,酯化反应中不添加酯化催化剂,因发明人发现酯化催化剂对酯化反应过程基本无影响。
[0070]
根据本发明,步骤1中,还加入助剂,助剂包括热稳定剂、抗氧剂等中的一种或几种,热稳定剂和抗氧剂能够防止聚酯热降解和氧降解。
[0071]
根据本发明,热稳定剂优选为磷酸三甲酯、亚磷酸三苯酯、磷酸三苯酯、亚磷酸三甲酯中的一种或几种,抗氧剂为抗氧剂1010或抗氧剂168,优选为抗氧剂1010。
[0072]
本发明中,在缩聚前加入热稳定剂如亚磷酸三苯酯有利于提高新型聚酯的色相。
[0073]
根据本发明,热稳定剂的加入量为tpa质量的0.1~0.5%,优选为0.2~0.4%。
[0074]
根据本发明,抗氧剂的加入量为tpa质量的0.1~0.5%,优选为0.2~0.4%。
[0075]
本发明中,步骤1得到酯化产物,酯化产物进行缩聚反应可得到自修复新型聚酯。
[0076]
步骤2、缩聚反应:将酯化产物进行缩聚反应,得到自修复新型聚酯。
[0077]
本发明中,在缩聚反应过程中需要加入缩聚催化剂来催化缩聚反应。
[0078]
本发明人发现,锑系催化剂对该缩聚反应没有催化效果,而钛系催化剂具有明显的催化效果,但钛系催化剂如钛酸四丁酯在投料前即与mpo和二甲酸单体同时加入时,无催化效果,可能是钛酸四丁酯在酯化反应时已经全部水解,且水解产物对缩聚反应无催化作用。
[0079]
根据本发明,缩聚催化剂为钛系催化剂,优选选自钛酸四丁酯、钛酸四乙酯、钛酸四正丙酯、钛酸四异丙酯或钛酸四异辛酯中的一种或几种,更优选为钛酸四丁酯(tbt)。
[0080]
本发明人还发现,在缩聚前直接加入钛系催化剂,由于设备和酯化平衡反应的特殊性,在酯化反应结束无水排除后仍有少量水在反应体系中或分馏柱中存在,反应体系中也会有少量二甲酸单体在抽真空时继续酯化生成水,这些水导致钛酸四丁酯水解,失去催化作用。
[0081]
根据本发明,在酯化反应结束后,将体系进行抽真空10-60min,尽量排除体系中的
水分,再加入缩聚催化剂,缩聚反应比较容易控制,缩聚反应时间也比较稳定。需要指出的是,在工业五釜流程(两酯化釜、两预缩聚釜和一终缩聚釜)生产聚酯时,缩聚催化剂也是在基本无水的第二预缩聚釜中加入,不会出现上述情况。
[0082]
本发明人发现,缩聚催化剂的添加量对缩聚反应速率、缩聚产品的色相及产品的副产物的含量均有较大影响。对缩聚反应速率直观的影响体现在缩聚时间上,缩聚催化剂的添加量较多时,聚合反应速率增加,聚合时间缩短,但可能导致产物发黄,缩聚催化剂添加量少,聚合反应速率低、聚合时间长。
[0083]
根据本发明,缩聚催化剂的加入量为tpa质量的0.1~1%,优选为0.3~0.8%,例如0.3%。
[0084]
根据本发明,步骤2中,在缩聚前还加入助剂,助剂包括热稳定剂、抗氧剂等中的一种或几种,热稳定剂和抗氧剂能够防止聚酯热降解和氧降解。
[0085]
根据本发明,热稳定剂优选为磷酸三甲酯、亚磷酸三苯酯、磷酸三苯酯、亚磷酸三甲酯中的一种或几种,抗氧剂为抗氧剂1010或抗氧剂168,优选为抗氧剂1010。
[0086]
本发明中,在缩聚前加入热稳定剂如亚磷酸三苯酯有利于提高自修复新型聚酯的色相,但抗氧剂的加入会增加所得自修复新型聚酯的黄度。本发明人发现,添加成核剂如纳米二氧化硅或纳米二氧化钛对聚酯的结晶无促进作用。
[0087]
根据本发明,热稳定剂的加入量为tpa质量的0.1~0.5%,优选为0.2~0.4%。
[0088]
根据本发明,抗氧剂的加入量为tpa质量的0.1~0.5%,优选为0.2~0.4%。
[0089]
本发明人发现,缩聚反应温度过高时反应速度并无明显加快,但副反应产物增加,产品颜色发黄,此外,缩聚反应温度与缩聚催化剂有关,催化效率高的催化剂,可适当降低缩聚反应温度,否则副产物增加,缩聚反应温度低,聚合物粘度增长较慢,即聚合反应速率较慢,聚合时间延长。
[0090]
根据本发明,缩聚反应温度为210~280℃,优选为210~260℃。
[0091]
根据本发明一种优选的实施方式,在2l的聚合反应釜中,缩聚反应温度为210~260℃,优选为230~260℃。
[0092]
根据本发明一种优选的实施方式,在30l的聚合反应釜中,缩聚反应温度为230~260℃,优选为240~255℃。
[0093]
本发明中,在缩聚反应中,真空度的高低对缩聚反应速度、缩聚反应温度和副产物均有影响,聚酯的缩聚反应是气相可逆反应,且平衡常数又很小,想要得到需要分子量的产品,就需要人为地打破反应平衡。聚酯缩聚反应的气相物质在生成物中,为了将促进缩聚反应的进行,采用抽真空降低生成物中气相产物的浓度是一个可行的办法。真空度的大小会影响反应的速率,真空度越大,越有利于气相产物的脱除,缩聚反应速率就越快。反之,不利于气相产物的脱除,缩聚反应速率变慢,甚至只能得到低聚物,无法得到足够高分子量的聚合物。但真空度过大会导致分子量分布变宽,也不利于聚合物的加工和产品的性能。
[0094]
根据本发明,步骤2中,缩聚反应的真空度为10~100pa,优选为10~80pa,优选为10~60pa。
[0095]
根据本发明,步骤2中,缩聚反应的时间包括反应体系未粘度未变化时间和粘度开始变化时间,反应体系粘度未变化(扭矩无变化)时间为10~60min,粘度开始变化(扭矩开始变化)到出料时间为20~180min。
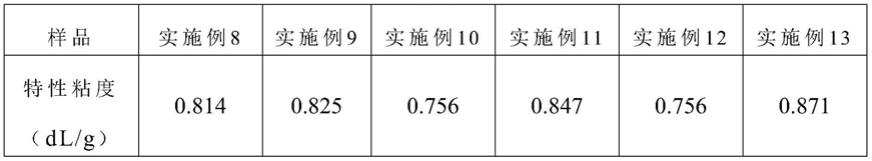
[0096]
根据本发明一种优选的实施方式,在2l的聚合反应釜中,反应体系粘度未变化时间为20~60min,粘度开始变化时间为30~90min。
[0097]
根据本发明一种优选的实施方式,在30l的聚合反应釜中,反应体系粘度未变化时间为10~20min,粘度开始变化时间为100~180min。
[0098]
根据本发明,当检测产物达到一定粘度后,缩聚反应结束,出料、切片,得到自修复新型聚酯。
[0099]
根据本发明,本发明的自修复新型聚酯的特性粘度大于0.7dl/g,优选可达到0.8dl/g以上,可用于制备聚酯纤维。
[0100]
根据本发明,所得自修复新型聚酯的mn为20000~30000,重均分子量mw为30000~40000,z均分子量为40000~50000,mw/mn为1.2~1.6,优选为1.3~1.6。
[0101]
根据本发明,测试所得自修复新型聚酯的流变性能,所得自修复新型聚酯为切力变稀的非牛顿流体,在同一剪切速率时,随着温度升高,熔体剪切粘度降低。
[0102]
根据本发明,所得自修复新型聚酯的玻璃化转变温度为40~60℃,无结晶峰和熔融峰,为非晶聚合物,初始热分解温度(热失重5%时的温度)为354~367℃,热分解残留率约为3~12%。
[0103]
本发明的自修复新型聚酯具有良好的形状恢复性,该自修复新型聚酯经过一定角度弯折后,经过55℃以上温度处理,形变结束后,弯折角度降低至1
°
~5
°
,恢复时间小于50s,形状恢复率高于88%;
[0104]
优选地,当经过60℃以上温度处理时,形变结束后,弯折角度降低至1
°
~3
°
,恢复时间小于8s,甚至达到1.5~5.8s,形状恢复率高于96%,更甚者为96.7~99.3%,形变恢复率能达到99.3%,基本恢复为变形前的形状。
[0105]
根据本发明,自修复新型聚酯经形变后,在60℃以上温度下处理,形变恢复性好,且恢复后具有良好的透过率,即恢复透明。
[0106]
本发明的自修复新型聚酯具备良好的可纺性,能够用于制备新型聚酯纤维。
[0107]
实施例
[0108]
实施例1
[0109]
该聚合反应在2l的聚合反应釜中进行,将500g对苯二甲酸、436g的2-甲基-1,3-丙二醇加入到反应釜中,酯化反应温度控制在237℃,酯化反应压力控制在270kpa,酯化反应加压时间120min,常压时间40min,出水量为74ml时,酯化反应结束,得到酯化产物;
[0110]
酯化反应结束后,抽真空20min,加入0.5ml tbt,缩聚反应压力30pa,控制缩聚反应温度260℃,缩聚反应时间90min(包括反应体系粘度未变化时间47min,以及粘度增长到出料时的时间43min),达到一定粘度后出料、切片,得到自修复新型聚酯。
[0111]
实施例2
[0112]
该聚合反应在2l的聚合反应釜中进行,将500g对苯二甲酸、414g的2-甲基-1,3-丙二醇加入到反应釜中,酯化反应温度控制在240℃,酯化反应压力控制在300kpa,酯化反应加压时间130min,常压时间60min,出水量为70ml时,酯化反应结束,得到酯化产物;
[0113]
酯化反应结束后,抽真空30min,加入0.7ml tbt,加入亚磷酸三苯酯1ml,控制缩聚反应压力60pa,控制缩聚反应温度260℃,缩聚反应时间150min(包括反应体系粘度未变化时间60min,以及粘度增长到出料时的时间90min),达到一定粘度后出料、切片,得到自修复
新型聚酯。
[0114]
实施例3
[0115]
该聚合反应在2l的聚合反应釜中进行,将500g对苯二甲酸、379g的二甲基-1,3-丙二醇加入到反应釜中,酯化反应温度控制在245℃,酯化反应压力控制在270kpa,酯化反应加压时间140min,常压时间40min,出水量为78ml时,酯化反应结束,得到酯化产物;
[0116]
酯化反应结束后,抽真空20min,加入1.0ml tbt,加入亚磷酸三苯酯1ml,缩聚反应压力10pa,控制缩聚反应温度260℃,缩聚反应时间80min(包括反应体系粘度未变化时间35min,以及粘度增长到出料时的时间45min),达到一定粘度后出料、切片,得到自修复新型聚酯。
[0117]
实施例4
[0118]
该聚合反应在2l的聚合反应釜中进行,将500g对苯二甲酸、379g的2-甲基-1,3-丙二醇加入到反应釜中,酯化反应温度控制在245℃,酯化反应压力控制在330kpa,酯化反应加压时间70min,常压时间50min,出水量为77ml时,酯化反应结束,得到酯化产物;
[0119]
酯化反应结束后,抽真空25min,加入1.5ml tbt,加入亚磷酸三苯酯1ml,缩聚反应压力10pa,控制缩聚反应温度260℃,缩聚反应时间57min(包括反应体系粘度未变化时间22min,以及粘度增长到出料时的时间35min),达到一定粘度后出料、切片,得到自修复新型聚酯。
[0120]
实施例5
[0121]
该聚合反应在2l的聚合反应釜中进行,将500g对苯二甲酸、352g的2-甲基-1,3-丙二醇加入到反应釜中,酯化反应温度控制在246℃,酯化反应压力控制在300kpa,酯化反应加压时间100min,常压时间40min,出水量为72ml时,酯化反应结束,得到酯化产物;
[0122]
酯化反应结束后,抽真空20min,加入1.5ml tbt,加入亚磷酸三苯酯1ml,加入抗氧剂0.5g,控制缩聚反应压力10pa,控制缩聚反应温度261℃,缩聚反应时间74min(包括反应体系粘度未变化时间29min,以及粘度增长到出料时的时间45min),达到一定粘度后出料、切片,得到自修复新型聚酯。
[0123]
实施例6
[0124]
该聚合反应在2l的聚合反应釜中进行,将500g对苯二甲酸、325g的2-甲基-1,3-丙二醇加入到反应釜中,酯化反应温度控制在240℃,酯化反应压力控制在300kpa,酯化反应加压时间120min,常压时间60min,出水量为67ml时,酯化反应结束,得到酯化产物;
[0125]
酯化反应结束后,抽真空15min,加入1.2ml tbt,加入亚磷酸三苯酯1ml,加入抗氧剂0.25g,控制缩聚反应压力10pa,控制缩聚反应温度259℃,缩聚反应时间73min(包括反应体系粘度未变化时间28min,以及粘度增长到出料时的时间45min),达到一定粘度后出料、切片,得到自修复新型聚酯。
[0126]
实施例7
[0127]
该聚合反应在2l的聚合反应釜中进行,将500g对苯二甲酸、325g的2-甲基-1,3-丙二醇加入到反应釜中,酯化反应温度控制在240℃,酯化反应压力控制在300kpa,酯化反应加压时间120min,常压时间60min,出水量为67ml时,酯化反应结束,得到酯化产物;
[0128]
酯化反应结束后,抽真空15min,加入1.2ml tbt,加入亚磷酸三苯酯1ml,加入抗氧剂0.25g,控制缩聚反应压力10pa,控制缩聚反应温度259℃,缩聚反应时间73min(包括反应
体系粘度未变化时间28min,以及粘度增长到出料时的时间45min),达到一定粘度后出料、切片,取间隔10min后的出料样品,得到自修复新型聚酯。
[0129]
实施例8
[0130]
该聚合反应在30l的聚合反应釜中进行,将3kg对苯二甲酸、2.11kg的2-甲基-1,3-丙二醇加入到反应釜中,酯化反应温度控制在242℃,酯化反应压力控制在300kpa,酯化反应加压时间240min,常压时间60min,出水率76.9%时,酯化反应结束,得到酯化产物;
[0131]
酯化反应结束后,抽真空60min,加入270ppm tbt,加入亚磷酸三苯酯1.5ml,控制缩聚反应压力30pa,控制缩聚反应温度255℃,缩聚反应时间115min(包括反应体系粘度未变化时间10min,以及粘度增长到出料时的时间105min),达到一定粘度后出料、切片,得到自修复新型聚酯。
[0132]
实施例9
[0133]
该聚合反应在30l的聚合反应釜中进行,将3kg对苯二甲酸、2.11kg的2-甲基-1,3-丙二醇加入到反应釜中,酯化反应温度控制在239℃,酯化反应压力控制在300kpa,酯化反应加压时间240min,常压时间60min,出水率69.2%时,酯化反应结束,得到酯化产物;
[0134]
酯化反应结束后,抽真空45min,加入270ppm tbt,加入亚磷酸三苯酯1.5ml,控制缩聚反应压力25pa,控制缩聚反应温度248℃,缩聚反应时间135min(包括反应体系粘度未变化时间15min,以及粘度增长到出料时的时间120min),达到一定粘度后出料、切片,得到自修复新型聚酯。
[0135]
实施例10
[0136]
该聚合反应在30l的聚合反应釜中进行,将3kg对苯二甲酸、2.11kg的2-甲基-1,3-丙二醇加入到反应釜中,酯化反应温度控制在247℃,酯化反应压力控制在300kpa,酯化反应加压时间180min,常压时间180min,出水率94.1%时,酯化反应结束,得到酯化产物;
[0137]
酯化反应结束后,抽真空20min,加入390ppm tbt,控制缩聚反应压力30pa,控制缩聚反应温度255℃,缩聚反应时间195min(包括反应体系粘度未变化时间15min,以及粘度增长到出料时的时间180min),达到一定粘度后出料、切片,得到自修复新型聚酯。
[0138]
实施例11
[0139]
该聚合反应在30l的聚合反应釜中进行,将3kg对苯二甲酸、2.11kg的2-甲基-1,3-丙二醇加入到反应釜中,酯化反应温度控制在245℃,酯化反应压力控制在300kpa,酯化反应加压时间140min,常压时间180min,出水率94.1%时,酯化反应结束,得到酯化产物;
[0140]
酯化反应结束后,抽真空20min,加入540ppm tbt,加入亚磷酸三苯酯1.5ml,控制缩聚反应压力30pa,控制缩聚反应温度255℃,缩聚反应时间135min(包括反应体系粘度未变化时间10min,以及粘度增长到出料时的时间125min),达到一定粘度后出料、切片,得到自修复新型聚酯。
[0141]
实施例12
[0142]
该聚合反应在30l的聚合反应釜中进行,将3kg对苯二甲酸、2.11kg的2-甲基-1,3-丙二醇、15g二氧化钛加入到反应釜中,酯化反应温度控制在245℃,酯化反应压力控制在300kpa,酯化反应加压时间180min,常压时间140min,出水率94.1%时,酯化反应结束,得到酯化产物;
[0143]
酯化反应结束后,抽真空20min,加入650ppm tbt,加入亚磷酸三苯酯1.5ml,控制
缩聚反应压力30pa,控制缩聚反应温度255℃,缩聚反应时间130min(包括反应体系粘度未变化时间10min,以及粘度增长到出料时的时间120min),达到一定粘度后出料、切片,得到自修复新型聚酯。
[0144]
实施例13
[0145]
该聚合反应在30l的聚合反应釜中进行,将3kg对苯二甲酸、3kg的2-甲基-1,3-丙二醇加入到反应釜中,酯化反应温度控制在247℃,酯化反应压力控制在300kpa,酯化反应加压时间150min,常压时间150min,出水率96.3%时,酯化反应结束,得到酯化产物;
[0146]
酯化反应结束后,抽真空20min,加入760ppm tbt,加入亚磷酸三苯酯1.5ml,控制缩聚反应压力30pa,控制缩聚反应温度255℃,缩聚反应时间190min(包括反应体系粘度未变化时间10min,以及粘度增长到出料时的时间180min),达到一定粘度后出料、切片,得到自修复新型聚酯。
[0147]
实验例
[0148]
实验例1
[0149]
对实施例1、3、5和7所得自修复新型聚酯进行红外测试,所得谱图如图1所示,图1中,曲线#1对应实施例1,曲线#3对应实施例3,曲线#5对应实施例5,曲线#7对应实施例7。
[0150]
从图1中可以看出,自修复新型聚酯样品均在1712cm-1
出现典型的酯基(-coo-)中羰基的伸缩振动峰(v-c=o
),说明形成了典型的酯化结构,且在红外谱图中未出现1700cm-1
附近的典型峰,端羧基量很少。
[0151]
实验例2
[0152]
特性粘度是聚合物相对分子量的代表参数之一,用来衡量聚合物相对分子量的大小,聚合物的相对分子量决定聚合物的用途。
[0153]
采用稀溶液粘度法测试实施例8-13所得自修复新型聚酯的特性粘度,测试条件为25℃,苯酚-四氯乙烷为溶剂,溶液浓度约为0.5g/100ml,毛细管粘度计(毛细管直径0.8mm)。
[0154]
采用“一点法”进行计算,测试结果如下表2所示。
[0155]
表2
[0156][0157]
从表2中可以看出,实施例8-13所得自修复新型聚酯的特性粘度大于0.7dl/g,甚至可达到0.847dl/g,可用于制备纤维。
[0158]
实验例3
[0159]
采用岛津15c高效液相色谱仪、rid-10a示差折光检测器、cto-20a柱温箱、岛津gpc色谱工作站测试实施例8-12所得mptt样品的分子量及分子量分布。
[0160]
液相色谱条件:流动相:四氢呋喃(thf)、流速流速:1ml/min,色谱柱:shodex gpc kf-803(8*300mm),柱温:40℃,进样体积:20μl。
[0161]
分别取实施例8-12的5个样品约10mg,溶于8ml四氢呋喃中,过夜。摇匀,用0.22μm
聚四氟膜过滤,待用。
[0162]
用2k、5k、10k、20k、40k peg标准样品绘制gpc校准曲线,线性回归方程为:y=-0.009727x3+0.2582x
2-2.6562x+13.4471。
[0163]
所得实施例gpc测试结果如表3所示,实施例8所得自修复新型聚酯的gpc色谱图和分子量分布视图如图2所示。
[0164]
表3
[0165]
样品mnmwmzmvmw/mn实施例123245327524251201.40899实施例225707366724792401.42655实施例327459372774734801.35757实施例421869335144496601.53248实施例524557369564844301.50487
[0166]
从表3中可以看出,所得自修复新型聚酯的mn在20000~30000范围内,mw在30000~40000范围内,mz在40000~50000范围内,mw/mn低于1.6,在1.3~1.6范围内。
[0167]
实验例4
[0168]
采用q2000型差示扫描量热仪(dsc,美国ta公司)测试试样的热稳定性,测试温度范围0~280℃,升温速度为10℃/min;采用209f1型热重分析仪(tg,德国耐驰公司)测试仪测试试样的热降解性能,测试条件为n2气氛,温度范围30~800℃,升温速率20℃/min。对实施例1-7所得自修复新型聚酯进行dsc和tg测试,图3为一次升温dsc曲线,图4为将实施例1和实施例2所得自修复新型聚酯样品在100℃下退火处理2h、4h和6h的dsc升温曲线,图5和图6为tg曲线,其中图6为图5中的tg曲线的局部放大图。在图3-6中,#1-实施例1,#2-实施例2,#3-实施例3,#4-实施例4,#5-实施例5,#6-实施例6,#7-实施例7。
[0169]
从图3中可以看出,样品dsc曲线上仅表现有玻璃化转变温度,在50℃左右,没有出现结晶峰,也未出现熔融峰,说明新型聚酯的结晶性很差,几乎不结晶,属于无法结晶的聚合物,可能与mpo结构中的侧甲基有关,图4中可与看出,样品在100℃下受热2-6h,仍然不能结晶,说明自修复新型聚酯几乎不结晶。
[0170]
从图5-6中可以看出,实施例1-7所得自修复新型聚酯的热降解失重过程的相差不大,样品的成炭性较差,按失重5%时的温度作为其初始分解温度(t
5wt%
),自修复新型聚酯样品的初始分解温度分别为354℃~367℃之间。
[0171]
实验例5
[0172]
研究自修复新型聚酯的流变性能以研究自修复新型聚酯的可纺性。对实施例10和13所得新型聚酯进行流变性能测试,测试自修复新型聚酯在不同温度时的剪切速率与剪切粘度的关系曲线。
[0173]
分别将实施例10和13所得自修复新型聚酯在75℃的真空干燥箱中干燥12h,冷却至室温,采用英国rosand rh10毛细孔流变仪对自修复新型聚酯进行不同温度的流变测试以获取不同温度下自修复新型聚酯的剪切速率和剪切粘度数据。测试结果如图7-8所示。图7为实施例10所得自修复新型聚酯在225℃、230℃和235℃下的剪切速率与剪切粘度关系。图8为实施例13所得自修复新型聚酯在225℃、230℃和235℃下的剪切速率与剪切粘度关系。
[0174]
从图7-8中可以看出,实施例10和实施例13所得自修复新型聚酯的剪切速率与剪切粘度的关系为切力变稀的非牛顿流体,在同一剪切速率时,随着温度升高,熔体剪切粘度降低。
[0175]
从图7-8中可以看出,剪切速率为1200s-1
时,235℃新型聚酯的剪切粘度约为170pa
·
s,因此自修复新型聚酯的可纺性试验可以235℃进行试验并根据纺丝状况进行调整。
[0176]
实验例6
[0177]
利用实施例1所得自修复新型聚酯制备直径3mm的圆柱型自修复新型聚酯样条,在常温下将自修复新型聚酯样条弯折一定角度θi,之后将弯折后的自修复新型聚酯样条放入不同温度(50℃、55℃、60℃、80℃和100℃)下的恒温水浴锅中,待形变恢复结束后,记录恢复时间,并采用量角器测定形变后的弯折角度θf,采用下式(1)计算形状恢复率rb,
[0178][0179]
式(1)中,当rb=1时,则规定具有完全的可形状恢复性;当rb=0时,则不具有形状恢复性。
[0180]
测试结果如表4-6所示,表4为θi为45
°
的自修复新型聚酯样条在不同水温下的弯折角度θf、恢复时间和形状恢复率,表5为θi为90
°
新型聚酯样条在不同水温下的弯折角度θf、恢复时间和形状恢复率,表6为θi为135
°
的自修复新型聚酯样条在不同水温下的弯折角度θf、恢复时间和形状恢复率。
[0181]
表4
[0182][0183]
表5
[0184]
[0185][0186]
表6
[0187][0188]
从表4-6中可以看出,自修复新型聚酯样条在50℃的水中不具有形变恢复性,当水温为55℃以上时,表现出形状可恢复性,但此时形变恢复后的弯折角度偏大、所需的恢复时间较长,恢复率偏低,且随着弯折角度越大,恢复时间变长。随着水温的升高,该自修复新型聚酯的形状恢复性能较好,恢复时间变短,100℃的恢复率达到99.3%,基本全部恢复为变形前的形状。
[0189]
将直径3mm的竖直的自修复新型聚酯样条弯折成英文字母“w”,作为变形前的样条状态,然后将样条置于60℃的恒温水浴锅中,采用照相机记录变形前和变形后的图片,分别如图9中(a)和(b)所示。
[0190]
从图9中可以看出,(a)自修复为新型聚酯的样条弯折后变成英文字母w的照片,从弯折处可看到应力发白现象,(b)为新型聚酯样条在60℃下恢复至变形前的竖直状态的照片,应力发白现象消失,自修复新型聚酯样条恢复透明,说明自修复新型聚酯样条在60℃下具有良好的形状恢复性,且还可恢复良好的透过率。
[0191]
以上结合优选实施方式和范例性实例对本发明进行了详细说明。不过需要声明的是,这些具体实施方式仅是对本发明的阐述性解释,并不对本发明的保护范围构成任何限制。在不超出本发明精神和保护范围的情况下,可以对本发明技术内容及其实施方式进行各种改进、等价替换或修饰,这些均落入本发明的保护范围内。本发明的保护范围以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1