一种达比加群酯中间体及其制备方法与流程
1.本发明属于药物化学技术领域,具体涉及一种达比加群酯及其关键中间体的合成方法。
背景技术:
2.达比加群酯化学名为β-丙氨酸,n-[[2-[[[4-[[[(己氧基)羰基]氨基]亚氨基甲基]苯基]氨基]甲基]-1-甲基-1h-苯并咪唑-5-基]羰基]-n-2-嘧啶-,己酯,甲磺酸盐;其化学结构如式i所示。是由勃林格殷格翰公司开发的口服直接凝血酶抑制剂,主要用于:(1)非瓣膜性房颤患者中风的预防;(2)已经过5-10天注射用抗凝药治疗的患者深静脉血栓(dvt)和肺栓塞(pe)预防;(3) 已经治疗的患者降低dvt和pe复发的风险;(4)髋关节置换术后dvt和pe的预防。
[0003][0004]
达比加群酯是一种新型的合成的直接凝血酶抑制剂,是达比加群的前体药物,属非肽类的凝血酶抑制剂。口服经胃肠吸收后,在体内转化为具有直接抗凝血活性的达比加群。达比加群结合于凝血酶的纤维蛋白特异结合位点,阻止纤维蛋白原裂解为纤维蛋白,从而阻断了血栓形成。达比加群可以从纤维蛋白-凝血酶结合体上解离,发挥可逆的抗凝作用。
[0005]
达比加群酯是直接凝血酶抑制剂,具有可以口服、强效、无需特殊用药监测、药物相互作用少等特点。体外、体内试验和临床各项研究均提示达比加群酯具有良好的疗效及药动学特性,临床应用前景乐观,其成功上市是抗凝血药物研究领域的一项重大突破。
[0006]
达比加群酯的制备方法首先在其化合物专利w09837075中公开,其制备方法为:原料4
-ꢀ
甲胺基-3-硝基苯甲酸(2)在氯化亚砜作用下形成4-甲胺基-3-硝基苯甲酰氯(3);3与3-(吡啶-2
-ꢀ
基氨基)丙酸乙酯(4)在三乙胺的存在下反应生成3-[(3-硝基-4-甲胺基苯甲酰基)(吡啶-2-基)氨基]丙酸乙酯(5);5经还原剂还原得到3-[(3-氨基-4-甲胺基苯甲酰基)(吡啶-2-基)氨基]丙酸乙酯(6);6先与n-(4-氰基苯基)氨基乙酸(7)在缩合剂的存在下形成酰胺,然后加入大过量乙酸回流得到3-[[[2-[[(4-氰基苯基)氨基]甲基]-1-甲基-1h-苯并咪唑-5-基]羰基]吡啶-2-基氨基]丙酸乙酯(8);将8溶于饱和的氯化氢乙醇溶液醇解生成3-[[[2-[[(4-乙氧基碳亚胺基苯基)氨基]甲基]-1-甲基-1h-苯并咪唑-5-基]羰基]
(吡啶-2-基)氨基]丙酸乙酯(9)的盐酸盐乙醇溶液,减压蒸馏出部分盐酸气后,再与氨或碳酸铵反应制得3-[[[2-[[(4-脒基苯基)氨基]甲基]-1-甲基-1h-苯并咪唑-5-基]羰基](吡啶-2-基)氨基]丙酸乙酯(10);10与氯甲酸正己酯在碱的作用下制得达比加群酯(11);11在丙酮溶液中与甲磺酸成盐制得达比加群酯甲磺酸盐。路线如下所示,该法主要存在以下问题:(1)3-[(3-氨基-4-甲胺基苯甲酰基)(吡啶-2-基)氨基]丙酸乙酯(6)与n-(4
-ꢀ
氰基苯基)氨基乙酸(7)缩合生成相应的苯并咪唑衍生物时,收率约50%,偏低,且需柱层析方式进行纯化;(2)用盐酸乙醇等试剂水解氰基得到脒基的反应操作较为繁琐,且会产生大量的废酸。
[0007][0008]
专利wo2011061080中对合成路线作了改进,如下所示:
[0009][0010]
该路线的重点是关键中间体3-丙氨酸-n-[[1-甲基-1h-苯并咪唑-2-卤甲基]-5-羰基]-n-2
-ꢀ
吡啶-乙基酯(16)的合成。中间体3-[(3-氨基-4-甲胺基苯甲酰基)(吡啶-2-基)氨基]丙酸乙酯(15)与偶合试剂发生环化缩合,得到关键中间体16。其中,所使用的偶合试剂为氯乙酸、氯乙酰氯、氯乙酸酐或三乙氧基氯乙烷。当使用氯乙酸时反应收率只有30%,且使用分子筛等控制体系为无水环境,反应条件苛刻;使用氯乙酰氯作偶合试剂时易产生二酰化副产物杂质,收率只有71%;氯乙酸酐价格较高,增加了生产成本;三乙氧基氯乙烷获得不易,需要自制,且制备较复杂。因此,该路线不适合中间体(16)和达比加群酯的工业化生产。
[0011]
专利cn102850325描述了达比加群酯关键中间体3-丙氨酸-n-[[1-甲基-1h-苯并咪唑-2-卤甲基]-5-羰基]-n-2-吡啶-乙基酯(16)的改良合成方法,如下所示:
[0012][0013]
此方法3-[(3-氨基-4-甲胺基苯甲酰基)(吡啶-2-基)氨基]丙酸乙酯(15)为原料,与偶合试剂得到中间体16,条件温和,反应收率比上述专利wo2011061080有所提高,但偶合剂价格较贵,不易获得。
[0014]
综上所述,虽然已有较多文献报道达比加群酯的合成方法,但这些方法存在明显缺陷,难以实现达比加群酯及中间体3-丙氨酸-n-[[1-甲基-1h-苯并咪唑-2-氯甲基]-5-羰基]-n-2-吡啶
ꢀ-
乙基酯的工业化规模制备。特别是针对中间体3-丙氨酸-n-[[1-甲基-1h-苯并咪唑-2-氯甲基]-5
-ꢀ
羰基]-n-2-吡啶-乙基酯的制备,目前还没有一种安全、环保、操
作性强且稳定可靠的方法。另一方面,为了确保临床用药的疗效和安全性,各国对药物中间体和原料药制定的质量标准越来越高,上述文献报道的达比加群酯的合成方法,均缺乏对工艺过程的研究和控制,没有建立完善的原料和中间体质量标准,无法保证终产品质量的稳定,难以直接应用于工业化制备达比加群酯。
技术实现要素:
[0015]
本发明所要解决的技术问题是提供一种反应条件温和、适合工业化生产的中间体3-丙氨酸-n-[[1-甲基-1h-苯并咪唑-2-氯甲基]-5-羰基]-n-2-吡啶-乙基酯及其制备方法,所得中间体纯度不低于99.0%。
[0016]
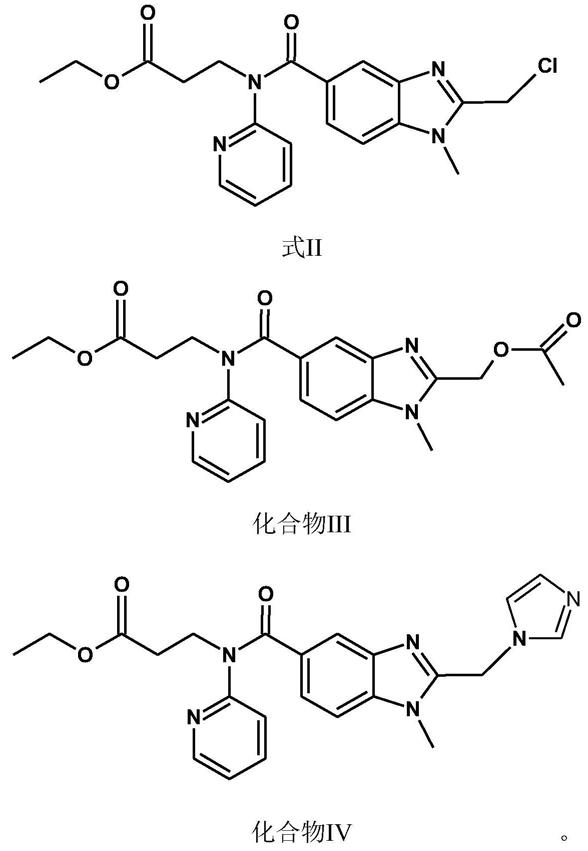
首先,本发明提供了一种达比加群酯中间体,化学名称为3-丙氨酸-n-[[1-甲基-1h-苯并咪唑-2-氯甲基]-5-羰基]-n-2-吡啶-乙基酯,结构如式ii所示,纯度不低于99.0%,其中化合物iii 的含量不高于0.5%,化合物iv的含量不高于0.3%。
[0017][0018][0019]
化合物iii、化合物iv为式(ii)中间体中的杂质,当其中化合物iii的含量不高于0.5%,化合物iv的含量不高于0.3%,在式(ii)中间体制备达比加群酯原料药的过程中,式(ii)中间体中化合物iii、化合物iv含量低,不会转移至终产品达比加群酯原料药中,因此不影响达比加群酯的原料药的质量。
[0020]
进一步的,所述的达比加群酯中间体,纯度不低于99.2%,其中化合物iii的含量不高于 0.4%,化合物iv的含量不高于0.3%。
[0021]
进一步的,所述的达比加群酯中间体,纯度不低于99.5%,其中化合物iii的含量不高于 0.3%,化合物iv的含量不高于0.2%。
[0022]
本发明进一步提供了上述达比加群酯中间体的制备方法。
[0023]
一种达比加群酯中间体3-丙氨酸-n-[[1-甲基-1h-苯并咪唑-2-氯甲基]-5-羰基]-n-2-吡啶
-ꢀ
乙基酯的制备方法,包括如下步骤:
[0024]
步骤a:反应釜1中,加入乙酸乙酯、3-[(3-氨基-4-甲胺基苯甲酰基)(吡啶-2-基)氨基] 丙酸乙酯,加热搅拌分散,降温20
±
10℃;
[0025]
步骤b:反应釜2中,加入乙酸乙酯、氯乙酸,控温20
±
10℃加入cdi,搅拌至体系澄清;
[0026]
步骤c:控温20
±
10℃,将反应釜2中反应液转移至反应釜1中;加入冰醋酸,升温至 40-60℃,反应至完全;
[0027]
步骤d:降温至20
±
5℃,加入纯化水,萃取分液,水相用乙酸乙酯萃取,合并乙酸乙酯相,减压浓缩至无馏分流出,得到油状物;
[0028]
步骤e:将步骤d得到的油状物,加入有机溶剂1,升温至40
±
5℃搅拌溶清,控温40
±ꢀ
5℃直接滴加有机溶剂2析晶,过滤,干燥,得到所述中间体。
[0029]
上述制备方法中,步骤a与步骤b不分先后顺序,步骤c、步骤d、步骤e按照先后顺序进行。
[0030]
上述制备方法中,所述的3-[(3-氨基-4-甲胺基苯甲酰基)(吡啶-2-基)氨基]丙酸乙酯,结构如式v所示:
[0031][0032]
所述的cdi为n,n-羰基-二咪唑,结构式如下所示:
[0033][0034]
上述制备方法中,以步骤a中3-[(3-氨基-4-甲胺基苯甲酰基)(吡啶-2-基)氨基]丙酸乙酯(式 v)的加入量为基准:
[0035]
步骤a乙酸乙酯的用量为2-10ml/g(体积重量比),优选5-7ml/g,更优选6ml/g。
[0036]
所述的步骤b中,乙酸乙酯的用量为2-10ml/g(体积重量比),优选3-5ml/g,更优选3.5-4.5 ml/g,更优选4ml/g。
[0037]
所述的步骤b中,氯乙酸的加入量为1.35-1.80摩尔当量(摩尔比,eq),优选1.35-1.65 摩尔当量(eq),更优选1.55摩尔当量(eq);所述的cdi的加入量为1.3-1.75摩尔当量(eq),优选1.3-1.6摩尔当量(eq),更优选1.5摩尔当量(eq)。
[0038]
所述的步骤c中,加入冰醋酸为转入完毕后立即加入冰醋酸。
[0039]
所述的步骤c中,冰醋酸的体积用量为4-10ml/g,优选4-8ml/g,更优选6ml/g。
[0040]
所述的步骤e中,所述的有机溶剂1选自丙酮、丙酮异丙醚的混合溶剂,所述的有机溶剂 2为异丙醚。
[0041]
所述的步骤e中,所述的有机溶剂1的体积用量为3-6ml/g,优选3-4ml/g,更优选3ml/g。
[0042]
所述的步骤e中,所述的有机溶剂2为体积用量为12-20ml/g,优选13-15ml/g。
[0043]
所述的步骤e中,有机溶剂1为丙酮异丙醚的混合溶剂,丙酮异丙醚的体积比为1:0.5-2;优选1:1。
[0044]
进一步的,本发明提供了一种检测3-丙氨酸-n-[[1-甲基-1h-苯并咪唑-2-氯甲基]-5-羰基]-n-2-吡啶-乙基酯、化合物iii、化合物iv的检测方法。
[0045]
一种3-丙氨酸-n-[[1-甲基-1h-苯并咪唑-2-氯甲基]-5-羰基]-n-2-吡啶-乙基酯、化合物iii、化合物iv纯度的检测方法,采用高效液相色谱法,色谱柱为c18色谱柱,4.6mm
×
250mm(直径
×
长度),5μm(粒径),检测波长为231nm,流动相a为ph3.8的乙酸铵缓冲液,流动相b 为乙腈,按下表进行梯度洗脱:
[0046]
时间流动相a(%)流动相b(%)067-7327-33567-7327-332557-6337-4325.167-7327-333067-7327-33
[0047]
进一步的,供试品加乙腈溶解,制备成含0.1-1mg/ml的溶液,注入液相色谱中。
[0048]
按照面积归一化法,计算中间体、化合物iii、化合物iv的纯度。
[0049]
使用本发明提供的达比加群酯中间体,制备得到的达比加群酯,纯度不低于99.5%,且不含有化合物iii、化合物iv。
附图说明:
[0050]
附图1:实施例2反应体系纯度色谱图
[0051]
附图2:实施例2中间体纯度色谱图
[0052]
附图3:实施例4中间体纯度色谱图
[0053]
附图4:实施例5中间体纯度色谱图
[0054]
附图5:对比例1反应体系纯度色谱图
[0055]
附图6:对比例3-1中间体纯度色谱图
[0056]
图谱中:
[0057]
vwd1b:紫外检测器;wavelength=231nm:检测波长;signal:信号
[0058]
rt[min]:保留时间[分钟];width[min]:峰宽;area:峰面积;area%:峰面积百分
比;
[0059]
height:峰高
[0060]
3-丙氨酸-n-[[1-甲基-1h-苯并咪唑-2-氯甲基]-5-羰基]-n-2-吡啶-乙基酯(式v)的出峰时间为13-14分钟;
[0061]
化合物iii的相对保留时间为0.68-0.78,即出峰时间为9.5-10.5分钟。
[0062]
化合物iv的相对保留时间为0.27-0.32,即出峰时间为3.8-4.2分钟。
具体实施方式
[0063]
本发明公开了一种达比加群酯中间体及其制备方法,本领域技术人员可以借鉴本发明的内容,结合药物化学的相关原理,适当改进工艺参数来实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明范围内。本发明的应用已经通过较佳实施例进行了描述,相关人员明显能在不脱离本发明内容、精神和范围内对本文所述的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。
[0064]
中间体的检测方法:
[0065]
色谱柱:xdb-c18(4.6
×
250mm,5μm),流速1.0ml/min
[0066]
流动相a:10mmol乙酸铵加乙酸调节ph至3.8
[0067]
流动相b:乙腈,按下表梯度进行洗脱,柱温40℃,进样量10μl,波长231nm
[0068]
时间ab070305703025604025.17030307030
[0069]
实施例1:达比加群酯中间体的制备
[0070]
步骤a:反应釜1中,加入乙酸乙酯600ml,式(v)化合物100g(0.29mol,1.0eq),加热至40
±
5℃搅拌分散,降温至20-30℃;
[0071]
步骤b:反应釜2中,加入乙酸乙酯400ml,氯乙酸43g(0.46mol,1.55eq),控温20
±
10℃加入cdi 71.2g(0.44mol,1.50eq),搅拌至体系澄清;
[0072]
步骤c:控温20
±
10℃,将反应釜2中反应液转移至反应釜1中;转入完毕后,立即加入冰醋酸600ml,10min内加入完毕,升温至50-60℃,保温反应3h,tlc跟踪反应完全;
[0073]
步骤d:降温至20
±
5℃,加入纯化水1000ml,萃取分液,水相用乙酸乙酯萃取,合并乙酸乙酯相,减压浓缩至无馏分流出;
[0074]
步骤e:将步骤d得到的油状物,加入丙酮150ml、异丙醚150ml的混合溶剂,升温至40
±ꢀ
5℃搅拌溶清,控温40
±
5℃,加入异丙醚1350ml,加入完毕后保温搅拌1h,自然降温至15
ꢀ±
5℃,保温析晶2h,过滤,干燥,得到式(ii)中间体化合物。中间体重89.2g,摩尔收率 76.0%。
[0075]
检测方法:取制备得到的中间体,加乙腈溶解,制备0.5mg/ml的溶液,按照中间体的检测方法测定,纯度99.62%,化合物iii 0.17%,化合物iv 0.14%,其它杂质0.07%。
[0076]
实施例2:达比加群酯中间体的制备
[0077]
步骤a:反应釜1中,加入乙酸乙酯300ml,式(v)化合物50g(0.15mol,1.0eq),加热至 40
±
5℃搅拌分散,降温至20-30℃;
[0078]
步骤b:反应釜2中,加入乙酸乙酯200ml,氯乙酸21.5g(0.23mol,1.55eq),控温20
±
10℃加入cdi 35.6g(0.22mol,1.50eq),搅拌至体系澄清;
[0079]
步骤c:控温20
±
10℃,将反应釜2中反应液转移至反应釜1中;转入完毕后,立即加入冰醋酸300ml,8min内加入完毕,升温至55-60℃,保温反应3.5h,tlc跟踪反应完全;将反应液用乙腈稀释,按照中间体的检测方法检测反应体系的纯度,中间体的纯度为93.83%,见附图1。
[0080]
步骤d:降温至20
±
5℃,加入纯化水550ml,萃取分液,水相用乙酸乙酯萃取,合并乙酸乙酯相,减压浓缩至无馏分流出;
[0081]
步骤e:将步骤d得到的油状物,加入丙酮150ml,升温至40
±
5℃搅拌溶清,控温40
±
5℃,加入异丙醚675ml,加入完毕后保温搅拌1h,自然降温至15
±
5℃,保温析晶2h,过滤,干燥,得到式(ii)中间体化合物。中间体重44.3g,摩尔收率75.5%。
[0082]
检测方法:取制备得到的中间体,加乙腈溶解,制备0.5mg/ml的溶液,按照中间体的检测方法测定,纯度99.59%,化合物iii 0.20%,化合物iv 0.14%,其它杂质0.08%,中间体色谱图见附图2。
[0083]
实施例3:达比加群酯中间体的制备
[0084]
步骤a:反应釜2中,加入乙酸乙酯800ml,氯乙酸84.5g(0.89mol,1.53eq),控温20
±
10℃加入cdi 142.5g(0.88mol,1.50eq),搅拌至体系澄清;
[0085]
步骤b:反应釜1中,加入乙酸乙酯1200ml,式(v)化合物200g(0.59mol,1.0eq),加热至 40
±
5℃搅拌分散,降温至20-30℃;
[0086]
步骤c:控温20
±
10℃,将反应釜2中反应液转移至反应釜1中;转入完毕后,立即加入冰醋酸1200ml,10min内加入完毕,升温至50-60℃,保温反应3h,tlc跟踪反应完全;
[0087]
步骤d:降温至20
±
5℃,加入纯化水1800ml,萃取分液,水相用乙酸乙酯萃取,合并乙酸乙酯相,减压浓缩至无馏分流出;
[0088]
步骤e:将步骤d得到的油状物,加入丙酮300ml、异丙醚300ml的混合溶剂,升温至40
±ꢀ
5℃搅拌溶清,控温40
±
5℃,加入异丙醚2700ml,加入完毕后保温搅拌1h,自然降温至15
ꢀ±
5℃,保温析晶2h,过滤,干燥,得到式(ii)中间体化合物。中间体重176.2g,摩尔收率 75.0%。
[0089]
检测方法:取制备得到的中间体,加乙腈溶解,制备0.5mg/ml的溶液,按照中间体的检测方法测定,纯度99.68%,化合物iii 0.15%,化合物iv 0.17%,其它杂质未检出。
[0090]
实施例4:达比加群酯中间体的制备
[0091]
步骤a:反应釜2中,加入乙酸乙酯210ml,氯乙酸22.1g(0.23mol,1.60eq),控温20
±
10℃加入cdi 35.6g(0.22mol,1.50eq),搅拌至体系澄清;
[0092]
步骤b:反应釜1中,加入乙酸乙酯330ml,式(v)化合物50g(0.15,1.0eq),加热至40
±
5℃搅拌分散,降温至20-30℃;
[0093]
步骤c:控温20
±
10℃,将反应釜2中反应液转移至反应釜1中;转入完毕后,立即加入冰醋酸300ml,8min内加入完毕,升温至45-55℃,保温反应2.8h,tlc跟踪反应完全;
[0094]
步骤d:降温至20
±
5℃,加入纯化水500ml,萃取分液,水相用乙酸乙酯萃取,合并乙酸乙酯相,减压浓缩至无馏分流出;
[0095]
步骤e:将步骤d得到的油状物,加入丙酮200ml,升温至40
±
5℃搅拌溶清,控温40
±
5℃,加入异丙醚675ml,加入完毕后保温搅拌1.5h,自然降温至15
±
5℃,保温析晶3h,过滤,干燥,得到式(ii)中间体化合物。中间体重43.2g,摩尔收率73.6%。
[0096]
取制备得到的中间体,加乙腈溶解,制备0.5mg/ml的溶液,按照中间体的检测方法测定,纯度99.22%,化合物iii 0.37%,化合物iv 0.19%,其它杂质0.23%,色谱图见附图3。
[0097]
实施例5:达比加群酯中间体的制备
[0098]
步骤a:反应釜1中,加入乙酸乙酯250ml,式(v)化合物50g(0.15,1.0eq),加热至40
±
5℃搅拌分散,降温至20-30℃;
[0099]
步骤b:反应釜2中,加入乙酸乙酯225ml,氯乙酸21.3g(0.23mol,1.54eq),控温20
±
10℃加入cdi 35.7g(0.22mol,1.50eq),搅拌至体系澄清;
[0100]
步骤c:控温20
±
10℃,将反应釜2中反应液转移至反应釜1中;转入完毕后,立即加入冰醋酸300ml,10min内加入完毕,升温至40-50℃,保温反应3.5h,tlc跟踪反应完全;
[0101]
步骤d:降温至20
±
5℃,加入纯化水600ml,萃取分液,水相用乙酸乙酯萃取,合并乙酸乙酯相,减压浓缩至无馏分流出;
[0102]
步骤e:将步骤d得到的油状物,加入丙酮50ml、异丙醚100ml的混合溶剂,升温至40
±
5℃搅拌溶清,控温40
±
5℃,加入异丙醚750ml,加入完毕后保温搅拌1h,自然降温至15
±
5℃,保温析晶2h,过滤,干燥,得到式(ii)中间体化合物。中间体重42.9g,摩尔收率73.1%。取制备得到的中间体,加乙腈溶解,制备0.5mg/ml的溶液,按照中间体的检测方法测定,纯度99.46%,化合物iii 0.32%,化合物iv 0.16%,其它杂质0.06%,色谱图见附图4。
[0103]
实施例6:达比加群酯中间体的制备
[0104]
步骤a:反应釜2中,加入乙酸乙酯175ml,氯乙酸21.2g(0.22mol,1.53eq),控温20
±
10℃加入cdi 35.7g(0.22mol,1.50eq),搅拌至体系澄清;
[0105]
步骤b:反应釜1中,加入乙酸乙酯350ml,式(v)化合物50g(0.15,1.0eq),加热至40
±
5℃搅拌分散,降温至20-30℃;
[0106]
步骤c:控温20
±
10℃,将反应釜2中反应液转移至反应釜1中;转入完毕后,立即加入冰醋酸300ml,10min内加入完毕,升温至50-60℃,保温反应2.5h,tlc跟踪反应完全;
[0107]
步骤d:降温至20
±
5℃,加入纯化水400ml,萃取分液,水相用乙酸乙酯萃取,合并乙酸乙酯相,减压浓缩至无馏分流出;
[0108]
步骤e:将步骤d得到的油状物,加入丙酮100ml、异丙醚50ml的混合溶剂,升温至40
±
5℃搅拌溶清,控温40
±
5℃,加入异丙醚600ml,加入完毕后保温搅拌1h,自然降温至15
±
5℃,保温析晶2h,过滤,干燥,得到式(ii)中间体化合物。中间体重42.5g,摩尔收率72.4%。取制备得到的中间体,加乙腈溶解,制备0.5mg/ml的溶液,按照中间体的检测方法测定,纯度99.33%,化合物iii 0.28%,化合物iv 0.19%,其它杂质0.20%。
[0109]
实施例7:考察步骤b氯乙酸、cdi的加入摩尔当量对达比加群酯中间体制备的影响
[0110]
步骤b中氯乙酸/cdi的加入量见下表,其余步骤和参数同实施例2;检测方法同实施例2。
[0111][0112]
氯乙酸、cdi的加入摩尔当量影响中间体的收率和纯度,氯乙酸的加入量为1.35-1.80摩尔当量(eq),cdi加入量1.3-1.75摩尔当量(eq)时,制备得到的中间体纯度高于99%,化合物iii的含量不高于0.5%,化合物iv的含量不高于0.3%。
[0113]
当氯乙酸加入量低于1.35eq且cdi的加入量低于1.3eq时(如实施例7-1、实施例7-2),制备得到的中间体摩尔收率明显下降,均低于61%,纯度低于99%,化合物iii的含量高于 0.5%。
[0114]
随着氯乙酸、cdi加入量的增加,收率并无增加,因此综合考虑成本,优选氯乙酸的加入量为1.35-1.65摩尔当量(eq),更优选1.55摩尔当量(eq);cdi加入量1.3-1.6摩尔当量(eq) 时,更优选1.5摩尔当量(eq)。
[0115]
实施例8:考察步骤c中醋酸的加入量对达比加群酯中间体制备的影响
[0116]
步骤c中醋酸的加入量见下表,其它步骤和参数同实施例2;检测方法同实施例2。
[0117][0118]
步骤c中醋酸的用量影响中间体的纯度,当醋酸用量较低时(对比例8-1中的2ml/g),制备得到的式(ii)中间体不仅收率下降,而且纯度较低(低于99%),化合物iv的含量较高(1.15%)。当醋酸用量为4-8ml/g时,制备得到的中间体纯度高于99%,化合物iii的含量不高于0.5%,化合物iv的含量不高于0.3%。
[0119]
对比例1:反应溶剂对达比加群酯中间体制备的影响
[0120]
以四氢呋喃代替步骤a和b中的乙酸乙酯,其余步骤及参数同实施例2;
[0121]
步骤c中反应体系的纯度,式(ii)中间体的纯度为84.84%,见附图5;
[0122]
步骤e所得中间体重量34.8g,摩尔收率59.3%,式(ii)中间体纯度93.83%。
[0123]
与乙酸乙酯相比,以四氢呋喃作为反应溶剂,存在以下两个问题:(1)以四氢呋喃为反应溶剂,步骤c完成后的反应体系中式(ii)中间体的纯度为84.84%。以乙酸乙酯为反应溶剂,实施例2步骤c完成后的反应液中纯度为93.83%,提示以四氢呋喃作为溶液,有大量的副产物生成,进而影响式(ii)中间体收率和纯度;(2)由于四氢呋喃与水部分互溶,结束后,萃取分液困难,需要用乙酸乙酯置换后再进行水洗分液操作,增加了操作复杂性和生产成本。而以乙酸乙酯作为反应溶剂,具有工艺简化、成本降低、收率提高、纯度提高等多方面优势。
[0124]
对比例2:步骤c中的冰醋酸的加入时间对达比加群酯中间体制备的影响
[0125]
改变步骤c中的冰醋酸的加入时间,用“转入完毕后,搅拌0.5h,加入冰醋酸300ml”代替实施例2中的“转入完毕后,立即加入冰醋酸300ml”,其余步骤及参数同实施2。
[0126]
步骤e所得式(ii)中间体重量41.8g,摩尔收率71.2%,式(ii)中间体纯度96.47%,化合物ⅲ0.42%,化合物ⅳ0.25%,杂质vi 2.59%,其他杂质0.27%。
[0127]
可见,不同的加料方式影响着中间体的纯度,尤其是步骤c冰醋酸的加入时间。实施例 2“将反应釜2中反应液转移至反应釜1中;转入完毕后,立即加入冰醋酸”,中间体纯度达到99.59%,且检测不到杂质vi。而对比例2“将反应釜2中反应液转移至反应釜1中;转入完毕后,搅拌0.5h,加入冰醋酸”,容易产生副产物杂质vi,得到的中间体中杂质vi含量明显增加,式(ii)中间体的纯度明显下降,仅有96.47%。
[0128][0129]
对比例3:步骤e中有机溶剂1、2的加入量对达比加群酯中间体制备的影响
[0130]
步骤e中有机溶剂1、有机溶剂2加入量见下表,其余步骤和参数同实施例2,检测方法同实施例2。
[0131][0132][0133]
步骤d得到的油状物溶解于异丙醚中,直接以异丙醚对油状物进行打浆纯化,得到中间体,此种方式在生产放大时存在较大的安全隐患,同时得到的式(ii)中间体的纯度为98.49%,低于99.0%(具体见附图6)。本技术发明人还发现:当有机溶剂1(丙酮或丙酮与异丙醚的混合溶剂量)低于3ml/g时(如对比例3-2中的1ml/g、对比例3-3中的2ml/g、对比例3-4及对比例3-5的2.5ml/g),并不能将油状物完全溶解,得到的中间体的纯度不理想,低于99%。
[0134]
对比例4:步骤c中反应温度对达比加群酯中间体制备的影响
[0135]
步骤c中反应温度见下表所示,其它步骤和参数同实施例2。
[0136][0137]
由对比例4及实施例1-8可知:步骤c加入冰醋酸后的反应温度影响中间体的纯度,反应温度较低时(对比例4-1中的30-40℃),反应3.5h后,tlc跟踪显示反应不完全,影响反应收率及所得的中间体的纯度;反应温度较高时(对比例4-2中的60-70℃),所得的中间体中化合物iii的含量增加,高于0.5%。因此反应温度优选40-60℃。
[0138]
实施例9:达比加群酯的制备
[0139]
1、酰脒的制备:
[0140][0141]
①
反应釜中加入纯化水、氢氧化钠,搅拌溶清;降温至5~15℃,加入丙酮,对氨基苯甲脒,搅拌至基本溶清;控温10~20℃,快速搅拌下滴加氯甲酸正己酯;滴毕保温反应1~2h,tlc 跟踪;
[0142]
②
分液,水相用乙酸乙酯萃取,合并有机相,用10%氯化钠水溶液反洗2次,有机相减压浓缩至无馏分流出;
[0143]
③
加入乙腈,搅拌溶清,控温0~10℃,快速搅拌下滴加浓盐酸,滴毕保温搅拌1~2h;过滤,干燥,得到酰脒。
[0144]
2、达比加群酯的制备
[0145][0146]
①
向反应釜中依次加入纯化水,碳酸氢钠,四氢呋喃,酰脒,搅拌10min,加入实施例3制备得到的中间体,nai,四氢呋喃,搅拌;将体系升温至50
±
5℃保温反应6
±
1h;
[0147]
②
反应完毕后降温至20
±
5℃,分液,水相用四氢呋喃萃取,合并有机相,用20%氯化钠水溶液~5v反洗;
[0148]
③
控温至20
±
5℃,向有机相中滴加正己烷,滴毕保温搅拌2h;过滤,得到达比加群
酯粗品。
[0149]
④
反应瓶中依次加入乙腈、达比加群酯粗品,加热至65
±
5℃溶清,降温至35-45℃,保温析晶1h,得达比加群酯(类白色固体,收率55%,纯度》99%)。
[0150]
3、甲磺酸达比加群酯的制备:
[0151]
室温依次加入丙酮和达比加群酯,35-45℃搅拌溶清,加入5%活性炭脱色30min,热抽滤,滤液降温至25-35℃;滴加甲磺酸~0.98eq的丙酮~2v溶液,滴加完毕后,搅拌,析出晶体,得到甲磺酸达比加群酯。
[0152]
得到达比加群酯,纯度为99.92%,不含化合物iii,化合物iv。
[0153]
结论:本发明方法制备的式(ⅱ)中间体所含化合物iii、化合物iv含量低,终产品达比加群酯中无化合物iii、化合物iv残留。
[0154]
实施例10:达比加群酯的制备
[0155]
1、酰脒的制备:
[0156][0157]
①
反应釜中加入纯化水、氢氧化钠,搅拌溶清;降温至5~15℃,加入丙酮,对氨基苯甲脒,搅拌至基本溶清;控温10~20℃,快速搅拌下滴加氯甲酸正己酯;滴毕保温反应1~2h,tlc 跟踪;
[0158]
②
分液,水相用乙酸乙酯萃取,合并有机相,用10%氯化钠水溶液反洗2次,有机相减压浓缩至无馏分流出;
[0159]
③
加入乙腈,搅拌溶清,控温0~10℃,快速搅拌下滴加浓盐酸,滴毕保温搅拌1~2h;过滤,干燥,得到酰脒。
[0160]
2、达比加群酯的制备
[0161][0162]
①
向反应釜中依次加入纯化水,碳酸氢钠,四氢呋喃,酰脒,搅拌10min,加入实施例7制备得到的中间体,nai,四氢呋喃,搅拌;将体系升温至50
±
5℃保温反应6
±
1h;
[0163]
②
反应完毕后降温至20
±
5℃,分液,水相用四氢呋喃萃取,合并有机相,用20%氯化钠水溶液~5v反洗;
[0164]
③
控温至20
±
5℃,向有机相中滴加正己烷,滴毕保温搅拌2h;过滤,得到达比加群酯粗品。
[0165]
④
反应瓶中依次加入乙腈、达比加群酯粗品,加热至65
±
5℃溶清,降温至35-45℃,保温析晶1h,得达比加群酯(类白色固体,收率55%,纯度》99%)。
[0166]
3、甲磺酸达比加群酯的制备:
[0167]
室温依次加入丙酮和达比加群酯,35-45℃搅拌溶清,加入5%活性炭脱色30min,热抽滤,滤液降温至25-35℃;滴加甲磺酸~0.98eq的丙酮~2v溶液,滴加完毕后,搅拌,析出晶体,得到甲磺酸达比加群酯。纯度为99.89%,不含化合物iii,化合物iv。
[0168]
结论:本发明方法制备的式(ⅱ)中间体所含化合物iii、化合物iv含量低,终产品达比加群酯中无化合物iii、化合物iv残留。
[0169]
实施例11:中间体的稳定性试验
[0170]
取实施例1、实施例7、实施例8中间体适量,在温度40℃
±
2℃;相对湿度75%
±
5%条件下放置6个月,分别于第0、3、6个月末取样,对有关物质等进行了测定。
[0171][0172]
本发明制备得到中的中间体质量稳定,长期贮存含量无明显变化。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1