一种泊马度胺中间体的合成方法与流程
本发明属于化学药物合成技术领域,涉及一种泊马度胺中间体的合成方法,具体涉及一种泊马度胺关键中间体3-氨基哌啶-2,6-二酮盐酸盐的合成方法。
背景技术:
泊马度胺,化学名称为3-氨基-n-(2,6-二氧代-3-哌啶基)邻苯二甲酰亚胺。2013年1月8日美国fda批准其治疗多发性骨髓瘤的患者。
多发性骨髓瘤起源于骨髓中的浆细胞,是一种恶性血液疾病,死亡率极高。临床主要表现为贫血、骨痛、肾损伤、高钙血症等,发病率为十万分之一,多发于50岁以上的老年人。
泊马度胺结构与沙利多胺类似,泊马度胺结构式如下:
泊马度胺具有抗肿瘤活性,可以抑制肿瘤细胞的增生分裂病诱导细胞凋亡。对治疗多发性骨髓瘤具有较好的疗效,具有广阔的市场。
泊马度胺可根据专利cn201410087575中所述的方法制备,以3-硝基邻苯二甲酸酐和本中间体3-氨基哌啶-2,6-二酮盐酸盐为原料,甲苯为溶剂,三乙胺为缚酸剂,n,n`-羰基二咪唑为缩合剂进行缩合反应,之后使用钯碳进行还原得到产物。
现有常用的合成方法主要是以谷氨酰胺为原料发生boc反应保护氨基,再与缩合剂反应环合,脱去保护基,与hcl成盐得到产物,合成路线如下。该路线不仅步骤长,而且使用到保护剂叔丁氧羰基和脱保护剂三氟乙酸,毒性大,尤其是三氟乙酸对骨的伤害较大。
技术实现要素:
本发明提供了泊马度胺关键中间体3-氨基哌啶-2,6-二酮盐酸盐的合成方法。旨在克服现有技术(以谷氨酰胺为原料发生boc反应保护氨基,再与缩合剂反应环合,脱去保护基,与hcl成盐得到产物)步骤长、原料毒性大、做出的产品纯度低,杂质多且成本高的缺点。此外,本发明方法具有条件温和,易控制,产品纯度高,杂质少且成本低廉的特点,是一条对环境友好的合成路线。
本发明是通过下述的技术方案来实现的:一种泊马度胺中间体的合成方法,所述泊马度胺中间体为3-氨基哌啶-2,6-二酮盐酸盐,结构式为
本发明提供的3-氨基哌啶-2,6-二酮盐酸盐的合成路线如下。
上述提到的各原料及产物分子式及名称如下:
所述的一种泊马度胺中间体的合成方法,包括以下步骤:
(1)l-谷氨酸在水中搅拌分散,滴加氨水直至继续搅拌反应0.5h;生成l-谷氨酸的二铵盐;
(2)加热,l-谷氨酸的二铵盐脱去一分子氨气环合生成3-氨基哌啶-2,6-二酮并在水中析出;
(3)过滤得到固体,溶于乙醇中,并向体系内通入盐酸气体,控制温度小于15℃,直至不再有固体析出;
(4)过滤,干燥得到产品3-氨基哌啶-2,6-二酮盐酸盐,收率可达82%,纯度98.7%。
步骤(1)中l-谷氨酸与氨水的摩尔比为2.0-2.5,优选1:2.5,水与l-谷氨酸的质量比为10:1。
步骤(1)中反应温度为温度在15-30℃,优选为15℃。
步骤(2)中反应温度为60-80℃,反应时间为2h,优选为60℃。
步骤(3)中乙醇用量为5-10倍质量的3-氨基哌啶-2,6-二酮,优选为10倍;反应温度小于15℃。
步骤(4)中干燥温度为70℃。
步骤(4)中干燥时间为3h。
本发明的有益效果在于:
(1)本发明方法操作简便、成本低廉,是一条对环境友好的合成路线。具体为其为泊马度胺的一种关键中间体。
(2)本发明采用原料l-谷氨酸和氨水反应生成l-谷氨酸的二铵盐,l-谷氨酸的二铵盐脱去一分子氨气环合生成3-氨基哌啶-2,6-二酮,溶于乙醇中,并向体系内通入盐酸气体,过滤干燥制得3-氨基哌啶-2,6-二酮盐酸盐。反应中未使用有毒、有害物质且单步反应简单易进行。不经过保护与脱保护,其原料较谷氨酰胺便宜,即保护环境又节约成本,且易控制。
附图说明:
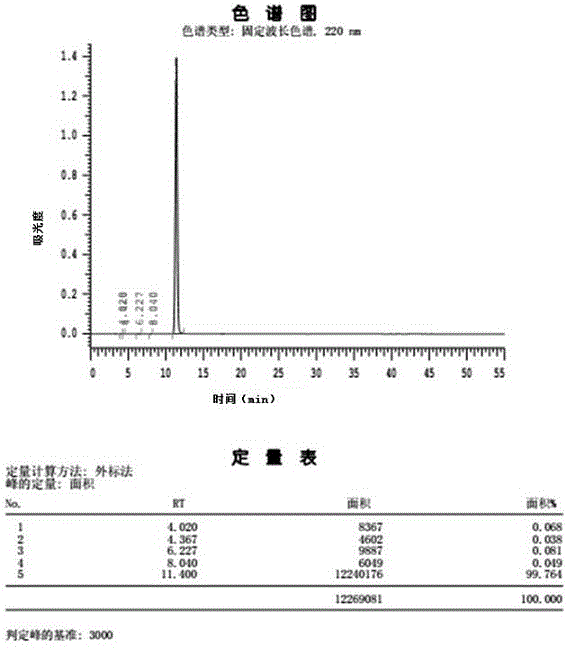
图1所示为实施例1产品液相色谱图。
具体实施方式
下面结合具体实施例对本发明作更进一步的说明,以便本领域的技术人员更了解本发明,但并不因此限制本发明。
本发明实施例中3-氨基哌啶-2,6-二酮盐酸盐的液相色谱检测条件为:
色谱条件:
色谱柱:ods2(250mm×4.6mm;5μm);
流速:0.8ml/min;
检测波长:220nm;
柱温:35℃;
稀释溶剂:50%乙腈盐溶液。
流动相:;0.03mkh2po4水溶液(磷酸调ph3.2)-乙腈(80:20).
流动相配制:
缓冲液500ml0.03mkh2po4盐水溶液(称取kh2po42.044g与水500ml混匀)以磷酸调ph3.2(ph=3.22),过滤,超声作为流动相。
流动相缓冲液400ml与100ml乙腈混合,即得。
稀释溶剂缓冲液100ml与100ml乙腈混合,即得。
溶液的制备
空白溶液:取稀释溶剂,即得。
供试品溶液的制备:取实施例1制得产品约10mg,精密称定,置25ml容量瓶中,加稀释溶剂溶解并稀释至刻度,摇匀,即得。
检测方法:精密量取上述供试品溶液10μl,注入液相色谱仪,记录色谱图,见说明书附图所示。
实施例1
(1)29.4gl-谷氨酸在294g水中搅拌分散,缓慢滴加8.75g氨水,控温小于15℃,溶清后继续保温搅拌反应0.5h,生成l-谷氨酸的二铵盐。
(2)加热至60℃反应2h,l-谷氨酸的二铵盐脱去一分子氨气环合生成3-氨基哌啶-2,6-二酮并在水中析出。
(3)过滤,105摄氏度恒温干燥3h,得3-氨基哌啶-2,6-二酮21.4g,将3-氨基哌啶-2,6-二酮21g溶于210g乙醇中,并向体系内通入盐酸气体,控制温度小于15℃,直至不再有固体析出。
(4)过滤,70℃干燥3h得到产品3-氨基哌啶-2,6-二酮盐酸盐27.0g,总收率82.1%,纯度99.76%。(该实施例所得产品用高效液相检测结果见图1)。
实施例2
(1)29.4gl-谷氨酸在294g水中搅拌分散,缓慢滴加7g氨水,控温小于15℃,溶清后继续保温搅拌反应0.5h,生成l-谷氨酸的二铵盐。
(2)加热至60℃反应2h,l-谷氨酸的二铵盐脱去一分子氨气环合生成3-氨基哌啶-2,6-二酮并在水中析出。
(3)过滤,105摄氏度恒温干燥3h,得3-氨基哌啶-2,6-二酮18g,3-氨基哌啶-2,6-二酮18g溶于180g乙醇中,并向体系内通入盐酸气体,控制温度小于15℃,直至不再有固体析出。
(4)过滤,70℃干燥3h得到产品3-氨基哌啶-2,6-二酮盐酸盐27.1g,总收率82.3%,纯度99.35%。
实施例3
(1)29.4gl-谷氨酸在294g水中搅拌分散,缓慢滴加8.75g氨水,控温小于15℃,溶清后继续保温搅拌反应0.5h,生成l-谷氨酸的二铵盐。
(2)加热至60℃反应2h,l-谷氨酸的二铵盐脱去一分子氨气环合生成3-氨基哌啶-2,6-二酮并在水中析出。
(3)过滤,105摄氏度恒温干燥3h,得3-氨基哌啶-2,6-二酮21.3g,3-氨基哌啶-2,6-二酮21.3g溶于213g乙醇中,并向体系内通入盐酸气体,控制温度30℃,直至不再有固体析出。
(4)过滤,70℃干燥3h得到产品3-氨基哌啶-2,6-二酮盐酸盐27.2g,总收率82.6%,纯度98.92%。
实施例4
(1)29.4gl-谷氨酸在294g水中搅拌分散,缓慢滴加8.75g氨水,控温小于15℃,容清后继续保温搅拌反应0.5h,生成l-谷氨酸的二铵盐。
(2)加热至80℃反应2h,l-谷氨酸的二铵盐脱去一分子氨气环合生成3-氨基哌啶-2,6-二酮并在水中析出。
(3)过滤,105摄氏度恒温干燥3h,得3-氨基哌啶-2,6-二酮18.7g,3-氨基哌啶-2,6-二酮18。7g溶于187g乙醇中,并向体系内通入盐酸气体,控制温度小于15℃,直至不再有固体析出。
(4)过滤,70℃干燥3h得到产品3-氨基哌啶-2,6-二酮盐酸盐27.1g,总收率82.3%,纯度99.12%。
- 还没有人留言评论。精彩留言会获得点赞!