白桦脂酸衍生物,其制备方法、药物组合物和应用
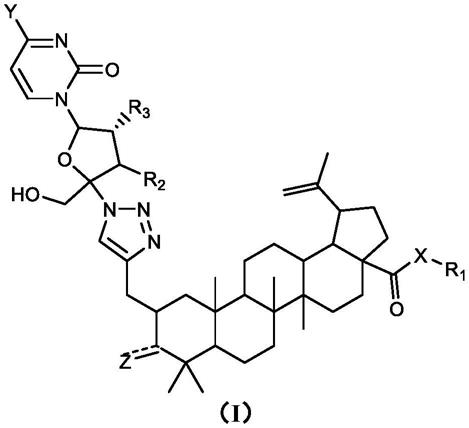
1.本发明属于医药领域,具体涉及白桦脂酸衍生物,其制备方法、药物组合物和应用。
背景技术:
2.白桦脂酸(betulinic acid,ba)是一种五环三萜类化合物,存在于白桦树、夏枯草、木瓜等多种植物中。研究发现,白桦脂酸具有广泛的生理活性,具有抗病毒、抗肿瘤、抗炎等效用。相较于白桦脂酸,其衍生物在抗肿瘤等方面具有更好的活性,如rpr103611和pa457(bevirimat)已经处于抗hiv的临床实验阶段,nvx-207在进行抗肿瘤的临床实验。但是由于其在水中的溶解性太差,导致生物利用度低及体内传输、代谢等方面的缺点。
技术实现要素:
3.为改善上述技术问题,本发明提供一种式(i)所示的白桦脂酸衍生物,其药学上可接受的盐、水合物或前药:
[0004][0005]
其中,x选自nra或o;
[0006]
r1选自h、无取代或任选被一个、两个或更多个rb取代的下列基团:c
1-40
烷基、c
3-20
环烷基、3-20元杂环基、c
6-20
芳基或5-20元杂芳基;
[0007]
y选自h、nrcr
c’、oh,无取代或任选被一个、两个或更多个rd取代的下列基团:c
1-40
烷基、c
3-20
环烷基、c
1-40
烷氧基、3-20元杂环基、c
6-20
芳基、5-20元杂芳基、3-20元杂环基氧基、c
6-20
芳基氧基或5-20元杂芳基氧基;
[0008]
r2、r3相同或不同,彼此独立地选自h、卤素、oh、sh、cn,无取代或任选被一个、两个或更多个re取代的下列基团:c
1-40
烷基或c
1-40
烷氧基;
[0009]
z选自为o、羟基或c
1-40
烷氧基,当为双键时,z为o;当其为单键时,z为oh或c
1-40
烷氧基;
[0010]
ra、rc、r
c’相同或不同,彼此独立地选自h、c
1-40
烷基、c
3-20
环烷基、3-20元杂环基、c
6-20
芳基;
[0011]
rb、rd、re相同或不同,彼此独立地选自卤素、oh、c
1-40
烷基、c
3-20
环烷基、c
1-40
烷氧基、3-20元杂环基、c
6-20
芳基、5-20元杂芳基、3-20元杂环基氧基、c
6-20
芳基氧基或5-20元杂芳基氧基。
[0012]
根据本发明的实施方案,所述白桦脂酸衍生物具有式(ii)所示的结构:
[0013][0014]
其中,x选自o;
[0015]
r1选自h或c
1-6
烷基;
[0016]
y选自nh2或oh;
[0017]
r2、r3相同或不同,彼此独立地选自h、f、cl、br、oh或sh;
[0018]
z选自为o、羟基或c
1-6
烷氧基,当为双键时,z为o;当其为单键时,z为oh或c
1-6
烷氧基。
[0019]
根据本发明的实施方案,所述白桦脂酸衍生物具有如下式(iii)所示的结构:
[0020][0021]
其中,x、y、z、r1、r2、r3独立地具有上文所述的定义。
[0022]
作为实例,所述白桦脂酸衍生物选自如下化合物:
[0023][0024]
本发明还提供所述白桦脂酸衍生物的制备方法,包括将化合物1与化合物2反应得到式(i)所示的白桦脂酸衍生物。
[0025][0026]
其中,x、y、z、r1、r2、r3独立地具有上文所述的定义。
[0027]
根据本发明的实施方案,所述反应可以在溶剂中进行;所述溶剂可以为水与有机溶剂的混合溶剂;所述有机溶剂可以为甲醇、乙醇、异丙醇、正丁醇、叔丁醇、四氢呋喃、二氧六环、n,n-二甲基甲酰胺;
[0028]
根据本发明的实施方案,所述水与有机溶剂的体积比为1:0.2-5,例如1:0.5-3,示例性为1:1;
[0029]
根据本发明的实施方案,所述反应在催化剂存在下进行,所述催化剂在反应过程中会形成一价铜离子,例如为cu和cuso4的组合、cucl、cui;
[0030]
根据本发明的实施方案,所述反应中化合物1与催化剂的摩尔比为1:0.1-1,例如为1:0.2-0.8;
[0031]
根据本发明的实施方案,所述反应中化合物1与化合物2的摩尔比为1:0.5-2,例如为1:0.8-1.2。
[0032]
本发明还提供一种药物组合物,其包含治疗有效量的所述白桦脂酸衍生物、其立
体异构体、氮氧化物、药学上可接受的盐。
[0033]
根据本发明的实施方案,所述药物组合物还包括一种或多种药学上可接受的辅料,例如载体或赋形剂。
[0034]
根据本发明的实施方案,所述药物组合物还可以进一步含有或不含有一种或多种额外的治疗剂。
[0035]
根据本发明的实施方案,所述药物组合物还包括赋形剂,通过所述赋形剂将药物组合物稀释或装入例如胶囊、小药囊、纸或其它容器形式的这种载体内。当赋形剂用作稀释剂时,它可以是固体、半固体或液体物质,用作溶媒、载体或活性成分的介质。因此,所述药物组合物可以是以下形式:片剂、丸剂、散剂、锭剂、小药囊、扁囊剂、酏剂、混悬剂、乳剂、溶液剂、糖浆剂、气雾剂(固体或溶于液体溶媒);例如含高达10%重量活性成分的软膏剂、软和硬明胶胶囊、栓剂、无菌注射溶液和无菌包装粉末。
[0036]
优选地,所述适宜赋形剂选自乳糖、葡萄糖、蔗糖、山梨醇、甘露醇、淀粉、阿拉伯胶、磷酸钙、藻酸盐、黄蓍胶、明胶、硅酸钙、微晶纤维素、聚乙烯吡咯烷酮、纤维素、水、糖浆和甲基纤维素。
[0037]
本发明还提供所述白桦脂酸衍生物,其立体异构体、氮氧化物、药学上可接受的盐、或前药中的至少一种在制备药物中的用途,其中所述药物为抗肿瘤药物。
[0038]
根据本发明的实施方案,所述肿瘤为癌。
[0039]
根据本发明的实施方案,所述药物可以用于抑制肿瘤细胞生长,所述肿瘤细胞可以为癌细胞,如选自宫颈癌细胞、肺腺癌细胞、乳腺癌细胞、肝癌细胞,其实例可以为选自下列的细胞:hela、c33a、siha、a549、mcf-7、luc7721。
[0040]
本发明还提供一种治疗肿瘤,例如癌症的方法,包括向有此需要的患者施用治疗有效量的所述白桦脂酸衍生物,其立体异构体、氮氧化物、药学上可接受的盐、或前药中的至少一种。
[0041]
根据本发明的实施方案,所述癌症可以为宫颈癌、肺腺癌、乳腺癌、肝癌。
[0042]
有益效果
[0043]
白桦脂酸室温下在水中溶解度仅为0.021μg/ml,水溶性很差。本发明通过对白桦脂酸的改造,使白桦脂酸和水溶性较好的活性核苷连接在一起,弥补了当前白桦脂酸类化合物水溶性差、生物利用率低以及核苷类化合物毒副作用大等问题。
[0044]
发明人意外地发现,本发明提供的手性化合物具备优异的抗肿瘤活性。并且,本发明提供合成方法简便可行,收率较高,将其应用于制备抗肿瘤的药物,具有较好的应用价值。
附图说明
[0045]
图1为化合物2的核磁共振氢谱。
[0046]
图2为化合物2的核磁共振碳谱。
[0047]
图3为化合物3的核磁共振氢谱。
[0048]
图4为化合物3的核磁共振碳谱。
[0049]
图5为化合物4的核磁共振氢谱。
[0050]
图6为化合物4的核磁共振碳谱。
[0051]
图7为化合物5的核磁共振氢谱。
[0052]
图8为化合物5的核磁共振碳谱。
[0053]
图9为化合物6的核磁共振氢谱。
[0054]
图10为化合物6的核磁共振碳谱。
[0055]
图11为化合物zx-6的核磁共振氢谱。
[0056]
图12为化合物zx-6的核磁共振碳谱。
[0057]
图13为化合物zx-1的核磁共振氢谱。
[0058]
图14为化合物zx-1的核磁共振碳谱。
[0059]
图15为化合物zx-2的核磁共振氢谱。
[0060]
图16为化合物zx-2的核磁共振碳谱。
[0061]
图17为化合物zx-3的核磁共振氢谱。
[0062]
图18为化合物zx-3的核磁共振碳谱。
[0063]
图19为化合物zx-4的核磁共振氢谱。
[0064]
图20为化合物zx-4的核磁共振碳谱。
[0065]
图21为化合物zx-5的核磁共振氢谱。
[0066]
图22为化合物zx-5的核磁共振碳谱。
[0067]
术语定义与说明
[0068]
除非另有说明,本技术说明书和权利要求书中记载的基团和术语定义,包括其作为实例的定义、示例性的定义、优选的定义、表格中记载的定义、实施例中具体化合物的定义等,可以彼此之间任意组合和结合。这样的组合和结合后的基团定义及化合物结构,应当属于本技术说明书记载的范围内。
[0069]“更多个”表示三个以上。
[0070]
术语“卤素”表示氟、氯、溴和碘。
[0071]
术语“c
1-40
烷基”应理解为表示具有1~40个碳原子的直链或支链饱和一价烃基。例如,“c
1-6
烷基”表示具有1、2、3、4、5或6个碳原子的直链和支链烷基。所述烷基是例如甲基、乙基、丙基、丁基、戊基、己基、异丙基、异丁基、仲丁基、叔丁基、异戊基、2-甲基丁基、1-甲基丁基、1-乙基丙基、1,2-二甲基丙基、新戊基、1,1-二甲基丙基、4-甲基戊基、3-甲基戊基、2-甲基戊基、1-甲基戊基、2-乙基丁基、1-乙基丁基、3,3-二甲基丁基、2,2-二甲基丁基、1,1-二甲基丁基、2,3-二甲基丁基、1,3-二甲基丁基或1,2-二甲基丁基等或它们的异构体。
[0072]
术语“c
3-20
环烷基”应理解为表示饱和的一价单环、双环烃环或桥环烷烃,其具有3~20个碳原子,优选“c
3-10
环烷基”。术语“c
3-10
环烷基”应理解为表示饱和的一价单环、双环烃环或桥环烷烃,其具有3、4、5、6、7、8、9或10个碳原子。所述c
3-10
环烷基可以是单环烃基,如环丙基、环丁基、环戊基、环己基、环庚基、环辛基、环壬基或环癸基,或者是双环烃基如十氢化萘环。
[0073]
术语“3-20元杂环基”意指饱和的一价单环、双环烃环或桥环烷烃,其包含1-5个独立选自n、o和s的杂原子的总成环原子数为3-20(如原子数为3、4、5、6、7、8、9、10等)的非芳族环状基团,优选“3-10元杂环基”。术语“3-10元杂环基”意指饱和的一价单环、双环烃环或桥环烷烃,其包含1-5个,优选1-3个选自n、o和s的杂原子。所述杂环基可以通过所述碳原子中的任一个或氮原子(如果存在的话)与分子的其余部分连接。特别地,所述杂环基可以包
基;噻吩基或亚噻吩基包括噻吩-2-基、亚噻吩-2-基、噻吩-3-基和亚噻吩-3-基;吡唑-1-基、吡唑-3-基、吡唑-4-基、吡唑-5-基。
[0077]
除非另有说明,本文中术语的定义同样适用于包含该术语的基团,例如c
1-40
烷基的定义也适用于c
1-40
烷氧基等。
[0078]
本领域技术人员可以理解,式i所示化合物可以以各种药学上可接受的盐的形式存在。如果这些化合物具有碱性中心,则其可以形成酸加成盐;如果这些化合物具有酸性中心,则其可以形成碱加成盐;如果这些化合物既包含酸性中心(例如羧基)又包含碱性中心(例如氨基),则其还可以形成内盐。酸加成盐包括但不限于:盐酸盐、氢氟酸盐、氢溴酸盐、氢碘酸盐、硫酸盐、焦硫酸盐、磷酸盐、硝酸盐、甲磺酸盐、乙磺酸盐、2-羟基乙磺酸盐、苯磺酸盐、甲苯磺酸盐、氨基磺酸盐、2-萘磺酸盐、甲酸盐、乙酰乙酸、丙酮酸、月硅酸酯、肉桂酸酯、苯甲酸盐、醋酸盐、二羟乙酸盐、三氟乙酸盐、三甲基乙酸盐、丙酸盐、丁酸盐、己酸盐、庚酸盐、十一酸盐、硬脂酸盐、抗坏血酸盐、樟脑酸盐、樟脑磺酸盐、柠檬酸盐、富马酸盐、苹果酸盐、马来酸盐、羟基马来酸盐、草酸盐、水杨酸盐、琥珀酸盐、葡萄糖酸盐、奎尼酸盐、双羟萘酸盐、甘醇酸盐、酒石酸盐、乳酸盐、2-(4-羟基苯甲酰基)苯甲酸盐、环戊烷丙酸盐、二葡糖酸盐、3-羟基-2-萘甲酸盐、烟酸盐、扑酸盐、果胶酯酸盐、3-苯基丙酸盐、苦味酸盐、特戊酸盐、衣康酸盐、三氟甲磺酸盐、十二烷基硫酸盐、对甲苯磺酸盐、萘二磺酸盐、丙二酸盐、己二酸盐、藻酸盐、扁桃酸盐、葡庚酸盐、甘油磷酸盐、磺基水杨酸盐、半硫酸或硫氰酸盐、天冬氨酸盐等;碱加成盐例如碱金属盐、碱土金属盐和铵盐等,具体包括但不限于:钠盐、锂盐、钾盐、铵盐、铝盐、镁盐、钙盐、钡盐、铁盐、亚铁盐、锰盐、亚锰盐、锌盐、铵盐(包括与nh3和有机胺形成的盐(nh4盐)、甲铵盐、三甲铵盐、二乙铵盐、三乙铵盐、丙铵盐、三丙铵盐、异丙铵盐、叔丁铵盐、n,n'-二苄基乙二铵盐、二环己铵盐、1,6-己二铵盐、苄铵盐、乙醇铵盐、n,n-二甲基乙醇铵盐、n,n-二乙基乙醇铵盐、三乙醇铵盐、氨丁三醇盐、赖氨酸盐、精氨酸盐、组氨酸盐、葡糖铵盐、n-甲基葡糖铵盐、二甲基葡糖铵盐、乙基葡糖铵盐、葡甲铵盐、甜菜碱盐、咖啡因盐、氯普鲁卡因盐、普鲁卡因盐、利多卡因盐、吡啶盐、甲基吡啶盐、哌啶盐、吗啉盐、哌嗪盐、嘌呤盐、可可碱盐、胆碱盐)等。
[0079]
根据其分子结构,本发明的化合物是手性的,因此可能存在各种对映异构体形式。因而这些化合物可以以消旋体形式或光学活性形式存在。本发明的化合物或其中间体可以通过本领域技术人员公知的化学或物理方法分离为对映异构体化合物,或者以此形式用于合成。在外消旋的胺的情况中,通过与光学活性的拆分试剂反应,从混合物制得非对映异构体。适当的拆分试剂的示例是光学活性的酸,例如r和s形式的酒石酸、二乙酰酒石酸、二苯甲酰酒石酸、扁桃酸、苹果酸、乳酸、适当的n-保护的氨基酸(例如n-苯甲酰脯氨酸或n-苯磺酰基脯氨酸)或各种光学活性的樟脑磺酸。借助光学活性的拆分试剂(例如固定在硅胶上的二硝基苯甲酰基苯基甘氨酸、三乙酸纤维素或其它碳水化合物的衍生物或手性衍生化的异丁烯酸酯聚合物),也可有利地进行色谱对映体拆分。用于此目的的适当的洗脱剂是含水或含醇的溶剂混合物,例如,己烷/异丙醇/乙腈。
[0080]
可以根据已知的方法,例如通过萃取、过滤或柱层析来分离相应的稳定异构体。
[0081]
术语“患者”是指包括哺乳动物在内的任何动物,优选小鼠、大鼠、其它啮齿类动物、兔、狗、猫、猪、牛、羊、马或灵长类动物,最优选人。
[0082]
本文中使用的短语“治疗有效量”是指研究人员、兽医、医师或其它临床医师正在
组织、系统、动物、个体或人中寻找的引起生物学或医学反应的活性化合物或药物的量。
具体实施方式
[0083]
下文将结合具体实施例对本发明的技术方案做更进一步的详细说明。应当理解,下列实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。凡基于本发明上述内容所实现的技术均涵盖在本发明旨在保护的范围内。
[0084]
除非另有说明,以下实施例中使用的原料和试剂均为市售商品,或者可以通过已知方法制备。
[0085]
实施例1:化合物的制备
[0086]
制备化合物zx-6采用如下方法:
[0087]
a.先用jones试剂将白桦脂酸氧化成白桦脂酮酸化合物2。
[0088]
b.使用草酰氯和甲醇将步骤a)制备的白桦脂酮酸化合物2酯化成白桦酯酮酸甲酯化合物3。
[0089]
c.使用溴代丙炔与步骤b)制备的白桦酯酮酸甲酯化合物3反应得到化合物4。
[0090]
d.采用硼氢化钠将步骤c)制备的化合物4中的羰基还原成羟基,制备得到化合物5。
[0091]
e.利用碘化锂将步骤d)中制备的化合物5脱去甲酯变为羧酸,制备得到化合物6.
[0092]
f.通过click反应将4
’-
叠氮胞苷加入到步骤e)制备的白桦脂酸化合物6中合成白桦脂酸衍生物zx-6。
[0093][0094]
具体合成路线如下:
[0095]
化合物2的合成:1000ml三口瓶中,加入白桦脂醇(20.0g,45.2mmol)和丙酮(400ml),然后在冰浴下滴加新制备的琼斯试剂(100ml),在冰浴下继续反应30分钟后撤去冰浴,室温搅拌8h,加入甲醇(250ml)和水(250ml)终止反应,蒸干溶剂,加入水,用乙酸乙酯萃取,合并有机相,干燥浓缩,柱色谱分离,得到白色固体2(12.2g,26.8mmol,59.3%)。1h nmr(dmso-d6,400mhz)δ:12.08(s,1h),4.70(s,1h),4.57(s,1h),2.90~2.99(m,1h),2.46~2.30(m,2h),2.25(dt,j=12.7,3.5hz,1h),2.15~2.08(m,1h),1.87~1.73(m,3h),1.65(s,3h),1.62(s,1h),1.54(t,j=11.3hz,1h),1.48~1.00(m,15h),0.98(s,3h),0.95(s,3h),0.93(s,3h),0.90(s,3h),0.85(s,3h)。
[0096]
化合物3的合成:在100ml圆底烧瓶中,将化合物2(4.54g,10mmol)溶于ch2cl2中,0℃滴加草酰氯(12.7g,100mmol)。反应完毕后蒸干溶剂,剩余物加入甲醇加热回流,tlc监测反应完毕后蒸干溶剂,加入乙酸乙酯萃取,水洗,有机相用na2so4干燥,柱色谱分离得到白色固体化合物3(3.36g,7.17mmol,71.7%)。1h nmr(cdcl3,400mhz)δ:4.74(s,1h),4.61(s,1h),3.68(s,3h),3.05~2.94(m,1h),2.56~2.32(m,2h),2.31~2.17(m,2h),1.96~1.83(m,3h),1.77~1.66(m,1h),1.69(s,3h),1.65~1.52(m,6h),1.51~1.11(m,13h),1.07(s,3h),1.02(s,3h),0.97(s,3h),0.95(s,3h),0.92(s,3h)。
13
c nmr(cdcl3,100mhz)δ:218.2,176.6,150.5,109.6,56.5,55.0,51.3,49.9,49.4,47.3,46.9,42.4,40.6,39.6,38.3,36.9,36.9,34.2,33.6,32.1,30.6,29.6,26.6,25.5,21.4,21.0,19.6,19.4,15.9,15.7,14.6。
[0097]
化合物4的合成:在500ml圆底烧瓶中,加入化合物3(2.54g,5.42mmol),和135ml四氢呋喃,室温搅拌加入1mol/l的kn(sime3)2的四氢呋喃溶液(40ml),搅拌1h后加入1mol/l三乙胺硼烷(net3b)的四氢呋喃溶液(40ml),继续搅拌1.5h,加入溴丙炔(4ml)。反应完毕后,加入3mol/l的hcl(3.0ml)淬灭反应,用乙酸乙酯萃取三次,合并有机相,将有机相用饱和碳酸氢钠溶液洗两次,na2so4干燥,柱色谱分离得到白色固体化合物4(2.32g,4.58mmol,84.5%)。1h nmr(cdcl3,400mhz)δ:4.72(s,1h),4.58(s,1h),3.67(s,3h),3.05~2.94(m,1h),2.91~2.77(m,1h),2.60(ddd,j=17.1,4.4,2.7hz,1h),2.33(dd,j=12.9,5.6hz,1h),2.29~2.10(m,3h),1.94(t,j=2.7hz,1h),1.91~1.83(m,1h),1.79~1.70(m,1h),1.67(s,3h),1.63~1.14(m,14h),1.17~1.01(m,3h),1.12(s,3h),1.06(s,3h),1.04(s,3h),0.98(s,3h),0.94(s,3h)。
13
c nmr(cdcl3,100mhz)δ:218.2,176.6,150.4,109.7,83.0,69.4,57.3,56.5,51.3,50.1,49.4,48.3,47.0,46.6,42.5,41.2,40.8,38.2,37.4,36.9,34.1,32.1,30.5,29.6,25.4,25.0,21.6,21.2,19.5,19.3,16.1,14.6。化合物5的合成:在500ml圆底烧瓶中,加入化合物4(2.03g,4.0mmol)、120ml甲醇和硼氢化钠(303mg,8.0mmol)。室温搅拌5h后,加入3mol/l的hcl(60.0ml),蒸干溶剂用乙酸乙酯萃取三次,合并有机相,将有机相用饱和碳酸氢钠溶液洗两次,将有机相干燥浓缩,柱色谱分离,用石油醚:乙酸乙酯=20:1做洗脱剂,得到白色固体化合物5(1.67g,3.28mmol,82.1%)。1h nmr(cdcl3,400mhz)δ:4.73(s,1h),4.59(s,1h),3.67(s,3h),3.07~2.93(m,2h),2.47~2.12(m,4h),1.98(t,j=2.4hz,1h),1.68(s,3h),1.58(t,j=11.4hz,3h),0.98(s,3h),0.97(s,3h),0.92(s,3h),0.86(s,3h),0.78(s,3h)。
13
c nmr(cdcl3,100mhz)δ:176.6,150.5,109.6,82.9,81.4,69.9,56.5,55.4,51.2,50.4,49.4,46.9,44.8,42.4,40.6,39.1,38.2,37.3,36.9,34.7,34.2,32.1,30.5,29.6,28.3,25.5,22.3,20.9,19.3,18.5,16.9,16.2,15.9,
d4,100mhz)δ:178.2,165.9,152.6,151.7,142.9,110.5,103.7,101.9,101.4,91.5,82.9,74.5,73.9,65.9,57.9,57.1,54.9,51.9,50.7,48.5,46.3,43.6,42.0,39.7,38.5,38.0,35.6,33.2,31.7,30.9,29.1,26.8,22.1,19.8,19.7,17.6,17.2,16.7,15.3.
[0102]
zx-3,白色固体,收率58%。1h nmr(meoh-d4,400mhz)δ:8.03(d,j=8.2hz,1h),7.92(s,1h),6.34(d,j=5.8hz,1h),5.79(d,j=8.1hz,1h),4.69(d,j=1.9hz,1h),4.65-4.54(m,3h),4.44(d,j=11.9hz,1h),3.98(d,j=11.9hz,1h),3.13(dd,j=14.4,3.0hz,1h),2.99(td,j=10.8,4.3hz,1h),2.81(d,j=10.8hz,1h),2.54(dd,j=14.3,8.9hz,1h),2.33-2.17(m,2h),2.00-1.84(m,3h),1.68(s,3h),0.98(s,3h),0.97(s,3h),0.93(s,3h),0.81(s,3h),0.80(s,3h),0.75-0.68(m,1h),0.61(t,j=12.7hz,1h).
13
c nmr(meoh-d4,100mhz)δ:180.1,166.0,152.6,152.0,146.3,142.9,123.9,110.3,103.7,101.0,91.4,82.9,74.5,74.0,66.0,57.5,57.1,52.0,50.5,48.6,46.2,43.7,42.0,40.5,39.7,38.5,38.2,37.3,35.6,33.4,31.8,30.9,29.7,29.1,26.9,22.1,19.8,19.7,17.5,17.1,16.7,15.2.
[0103]
zx-4,白色固体,收率72%。1h nmr(meoh-d4,400mhz)δ:7.98(d,j=7.5hz,1h),7.94(s,1h),6.29(d,j=5.5hz,1h),5.97(brs,1h),4.70(s,1h),4.66-4.53(m,3h),4.41(d,j=12.0hz,1h),3.99(d,j=11.9hz,1h),3.66(s,3h),3.24-3.09(m,2h),2.98(td,j=10.6,4.5hz,1h),2.59(dd,j=14.1,6.8hz,1h),2.30-2.16(m,2h),2.06(dd,j=12.6,5.2hz,1h),1.93-1.78(m,2h),1.68(s,3h),1.12(s,3h),1.07(s,3h),1.04(s,3h),0.99(s,3h),0.98(s,3h).
13
c nmr(meoh-d4,100mhz)δ:218.6,178.2,167.9,158.6,151.8,146.2,143.8,124.1,110.5,100.9,96.9,93.5,74.8,73.9,66.0,58.9,58.0,51.9,51.5,50.7,49.6,48.6,48.3,43.7,43.6,42.1,39.7,38.8,37.9,35.4,33.2,31.7,30.9,27.1,26.8,25.7,22.3,22.1,20.5,19.6,16.8,16.6,15.2.
[0104]
zx-5,白色固体,收率70%。1h nmr(meoh-d4,400mhz)δ:8.01(d,j=7.5hz,1h),7.97(s,1h),6.33(d,j=5.2hz,1h),6.01(brs,1h),4.69(d,j=1.7hz,1h),4.66-4.53(m,3h),4.43(d,j=11.9hz,1h),4.00(d,j=11.9hz,1h),3.64(s,3h),3.13(d,j=11.7hz,1h),2.97(td,j=10.9,4.8hz,1h),2.82(d,j=10.7hz,1h),2.56(dd,j=14.1,8.8hz,1h),2.28-2.14(m,2h),2.01-1.80(m,3h),1.68(s,3h),0.97(s,6h),0.90(s,3h),0.81(s,3h),0.80(s,3h),0.75-0.65(m,1h),0.61(t,j=12.6hz,1h).
13
c nmr(meoh-d4,100mhz)δ:178.2,167.6,151.8,146.3,143.7,124.0,110.4,101.1,93.4,82.9,74.8,73.9,66.0,58.0,57.0,52.0,51.9,50.7,48.6,46.2,43.6,42.0,40.5,39.7,38.5,38.0,37.3,35.6,33.2,31.7,30.9,29.8,29.1,26.9,22.1,19.8,19.7,17.5,17.1,16.6,15.3.
[0105]
实施例2:生物活性测试
[0106]
2.1本发明化合物对hela、c33a、siha细胞的抑制活性测试
[0107]
本实验用不同浓度的受试化合物分别作用于人宫颈癌细胞系hela、c33a、siha细胞48小时,通过mtt法检测细胞生长抑制率,来检测化合物对不同肿瘤细胞的抑制程度。
[0108]
实验方法:
[0109]
将受试溶解于一定体积的dmso中,配成浓度为10mmol/l的母液4℃保存,使用的时候取母液稀释为一定倍数的溶液。
[0110]
具体操作步骤如下:
[0111]
(1)取处于对数生长期的hela、c33a、siha细胞,离心、重悬,调整细胞悬液密度至8
×
104cell/ml,接种于96孔培养板中,每孔加入100μl细胞悬液,然后放置细胞培养箱中37℃,5%co2常规培养。
[0112]
(2)孵育24h后,显微镜下观察各孔细胞单层贴壁均匀生长,实验组孔内分别加入不同浓度的受试化合物溶液1μl,使化合物终浓度均分别为1.5、3、6.25、12.5、25、50、75、100μmol/l,每个浓度均设4个复孔;同时设空白组(只加入培养基不加细胞悬液)、阴性对照组(只加入细胞悬液)。
[0113]
(3)放置培养箱培养48h后,每孔加入20μl mtt溶液,在培养箱中继续培养4h。然后小心弃去上层培养基,每孔加入150μl dmso,至于摇床上37℃恒温震荡10min,使结晶物充分溶解。在酶联免疫检测仪490nm波长下检测各孔吸光度od值。
[0114]
(4)计算样品对细胞的抑制作用,公式如下:
[0115]
抑制率(%)=(细胞对照孔od值-给药孔od值)/(细胞对照孔od值-空白孔od值)
×
100%
[0116]
根据所计算的抑制率,求出各受试化合物抑制细胞生长的半数抑制浓度ic
50
值。
[0117]
表1:受试化合物的ic
50
值(μmol/l)
[0118][0119]
由表1可以看出:一定剂量的zx-1~zx-6样品均对人宫颈癌细胞存在不同程度的抑制作用;zx-6对于宫颈癌细胞的抑制作用较好,ic
50
值较低。
[0120]
2.2本发明化合物对a549、mcf-7和luc7721细胞的抑制活性测试
[0121]
本实验用不同浓度的受试化合物分别作用于a549、mcf-7和luc7721细胞48小时,通过mtt法检测细胞生长抑制率,来检测化合物对不同肿瘤细胞的抑制程度。
[0122]
实验方法:
[0123]
将受试化合物溶解于一定体积的dmso中,配成浓度为10mmol/l的母液4℃保存,使用的时候取母液稀释为一定倍数的溶液。
[0124]
具体操作步骤如下:
[0125]
(1)取处于对数生长期的a549、mcf-7和luc7721细胞,离心、重悬,调整细胞悬液密度至8
×
104cell/ml,接种于96孔培养板中,每孔加入100μl细胞悬液,然后放置细胞培养箱中37℃,5%co2常规培养。
[0126]
(2)孵育24h后,显微镜下观察各孔细胞单层贴壁均匀生长,实验组孔内分别加入不同浓度的受试化合物溶液1μl,使化合物终浓度均分别为1.5、3、6.25、12.5、25、50、75、100μmol/l,每个浓度均设4个复孔;同时设空白组(只加入培养基不加细胞悬液)、阴性对照组(只加入细胞悬液)。
[0127]
(3)放置培养箱培养48h后,每孔加入20μl mtt溶液,在培养箱中继续培养4h。然后小心弃去上层培养基,每孔加入150μl dmso,至于摇床上37℃恒温震荡10min,使结晶物充分溶解。在酶联免疫检测仪490nm波长下检测各孔吸光度od值。
[0128]
(4)计算样品对细胞的抑制作用,公式如下:
[0129]
抑制率(%)=(细胞对照孔od值-给药孔od值)/(细胞对照孔od值-空白孔od值)
×
100%。
[0130]
根据所计算的抑制率,求出各受试化合物抑制细胞生长的半数抑制浓度ic
50
值。
[0131][0132]
表2:受试化合物的ic
50值
(μmol/l)
[0133][0134]
由表2可以看出:本发明实施例化合物具有更优异的抗癌活性。
[0135]
以上,对本发明的实施方式进行了说明。但是,本发明不限定于上述实施方式。凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1