一种高纯度达比加群酯及其制备方法和其应用与流程
1.本发明属于医药化工领域,具体涉及一种高纯度达比加群酯及其制备方法和其应用。
背景技术:
2.达比加群酯为非肽类的凝血酶抑制剂。口服经胃肠吸收后,在体内转化为具有直接抗凝血活性的达比加群。达比加群结合于凝血酶的纤维蛋白特异结合位点,阻止纤维蛋白原裂解为纤维蛋白,从而阻断了凝血瀑布网络的最后步骤及血栓形成。并且,达比加群可从纤维蛋白—凝血酶结合体上解离而发挥可逆的抗凝作用。
3.wo2014/192030公开了一种达比加群酯的制备方法,包括下述步骤:
[0004][0005]
但该方法制得的formula 5化合物中所含杂质a含量较高,杂质a将在后续达比加群酯的制备中衍生产生与达比加群酯结构相近的杂质b,而杂质b通过常规重结晶等方法无法实现有效纯化,即使在最后制备达比加群酯甲磺酸盐步骤仍无法纯化去除且衍生成杂质c(从而易导致衍生杂质c含量超标的问题),进而影响达比加群酯甲磺酸盐的纯度和质量,杂质c含量远超过《化学药物杂质研究技术指导原则》规定的限度(≤0.1%),无法达到注册上市标准,给企业造成不可估量的损失。为此,急需开发一种高纯度的达比加群酯制备方法。
[0006]
技术实现要素:
[0007]
本发明的目的在于提供一种高纯度的达比加群酯或其药学上可接受的盐,所述达比加群酯的纯度不低于99.0%,且其中杂质b的含量不超过0.1%。
[0008][0009]
本发明的优选技术方案中,所述达比加群酯或其药学上可接受的盐的纯度不低于99.2%,优选不低于99.4%。
[0010]
本发明的优选技术方案中,所述达比加群酯或其药学上可接受的盐中杂质b的含量不超过0.05%,优选不超过0.03%。
[0011]
本发明的优选技术方案中,所述达比加群酯药学上可接受的盐中杂质c的含量不超过0.1%,优选不超过0.05%,再优选不超过0.03%。
[0012]
本发明的优选技术方案中,所述达比加群酯药学上可接受的盐选自其对甲苯磺酸盐、甲磺酸盐、磺酸盐、盐酸盐、氢溴酸盐、硫酸盐、磷酸盐、硝酸盐、酒石酸盐、富马酸盐、马来酸盐、柠檬酸盐、乙酸盐、甲酸盐、苯甲酸盐、肉桂酸盐、丁二酸盐、丙二酸盐中的任一种或其组合。
[0013]
本发明的另一目的在于提供一种高纯度的式ⅰ所示结构的中间体或其药学上可接受的盐,所述式ⅰ所示结构的中间体或其药学上可接受的盐的纯度不低于97%,且其中杂质a的含量不超过1.0%。
[0014][0015]
本发明的优选技术方案中,所述式ⅰ所示结构的中间体或其药学上可接受的盐中的杂质a含量不超过0.8%,优选不超过0.6%,更优选不超过0.5%。
[0016]
本发明的优选技术方案中,所述式ⅰ所示结构的中间体的药学上可接受的盐选自其对甲苯磺酸盐、甲磺酸盐、磺酸盐、盐酸盐、氢溴酸盐、硫酸盐、磷酸盐、硝酸盐、酒石酸盐、富马酸盐、马来酸盐、柠檬酸盐、乙酸盐、甲酸盐、苯甲酸盐、肉桂酸盐、丁二酸盐、丙二酸盐中的任一种。
[0017]
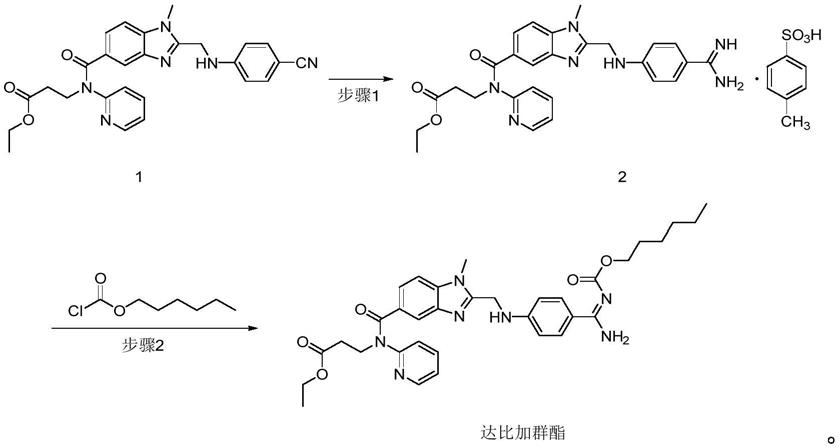
本发明的另一目的在于提供一种达比加群酯的制备方法,包括下述步骤:(1)化合物1进行pinner反应后与对甲苯磺酸成盐,制得化合物2;(2)将化合物2与氯甲酸己酯发生反应,制得达比加群酯。
[0018][0019]
本发明的优选技术方案中,所述步骤(1)包括下述步骤:化合物1与hcl/乙醇、对甲苯磺酸或其一水合物和氨水进行反应;将所得反应混合物冷却析出固体,过滤,滤饼处理制得化合物2粗品,纯化,制得化合物2的纯化品。
[0020]
本发明的优选技术方案中,所述步骤(1)中的滤饼处理包括下述步骤:滤饼室温下置于水中搅拌,分离,收集固体;再将所述固体在室温下置于乙醇中搅拌,室温或降温析出固体,干燥,即得。
[0021]
本发明的优选技术方案中,所述步骤(1)纯化中,将制得的化合物2粗品加入到纯化溶剂中,搅拌,分离,收集固体,淋洗,干燥,即得。
[0022]
本发明的优选技术方案中,所述纯化溶剂选自dmf、h2o、丙酮、四氢呋喃、dmso、乙醇中的任一种或其组合。
[0023]
本发明的优选技术方案中,所述dmf/h2o组合中,dmf:h2o的体积比为1:1-1:10,优选为1:2-1:8,更优选为1:3-1:6,再优选为1:3.5-1:4.5。
[0024]
本发明的优选技术方案中,所述丙酮/dmso组合中,丙酮:dmso的体积比为2-3:1,
优选为1.5:1。
[0025]
在本发明的优选技术方案中,所述步骤(1)纯化中,化合物2粗品与纯化溶剂的质量体积比为1:3-20,优选为1:5-15。
[0026]
本发明的优选技术方案中,所述步骤(1)纯化中,所述分离选自过滤、离心、膜处理的任一种或其组合。
[0027]
本发明的优选技术方案中,所述步骤(1)纯化中,淋洗溶剂选自dmf、乙醇、丙酮、四氢呋喃、水中的任一种或其组合。
[0028]
本发明的优选技术方案中,所述步骤(1)纯化中,化合物2粗品与淋洗溶剂的质量体积比为1:1-10,优选为1:2-5。
[0029]
本发明的优选技术方案中,所述步骤(1)纯化中,所述干燥选自真空干燥、减压干燥、常压干燥、喷雾干燥、沸腾干燥的任一种或其组合。
[0030]
本发明的优选技术方案中,所述步骤(1)纯化中,干燥温度为25-80℃,优选为35-65℃,更优选为40-50℃。
[0031]
本发明的优选技术方案中,所述步骤(1)制备所得化合物2中,杂质a含量不超过0.8%,优选不超过0.6%,更优选不超过0.5%。
[0032]
本发明的优选技术方案中,所述步骤(2)制备所得达比加群酯的纯度不低于99.0%,优选纯度不低于99.2%,更优选纯度不低于99.4%。
[0033]
本发明的优选技术方案中,所述步骤(2)制备所得达比加群酯中的杂质b含量不超过0.1%,优选不超过0.05%,再优选不超过0.03%。
[0034]
本发明的另一目的在于提供本发明的高纯度达比加群酯用于制备用于预防和/或治疗血栓性疾病的药物中的应用。
[0035]
本发明的优选技术方案中,所述血栓性疾病选自动脉血栓性疾病或静脉血栓性疾病。
[0036]
本发明的优选技术方案中,所述血栓性疾病选自下肢深静脉血栓形成、搭桥手术或血管成形术(pt(c)a)后再次闭塞、周围动脉疾病闭塞、肺栓塞、弥散性血管内凝血、冠状动脉血栓形成、中风以及分流器或支架闭塞中的任一种或其并发症。
[0037]
本发明的另一目的在于提供一种药物组合物,所述药物组合物含有本发明的高纯度达比加群酯和p-gp抑制剂、p-gp诱导剂、p-gp底物药物、非甾体抗炎药、质子泵抑制剂、h2受体抑制剂、抗凝血药物中的一种或其组合。
[0038]
本发明的优选技术方案中,所述p-gp抑制剂选自酮康唑、伊曲康唑、决奈达隆、胺碘酮、奎尼丁、维拉帕米、克拉霉素、非洛地平、尼卡地平、硝苯地平、尼群地平、他克莫司、米非司酮、特非那定、环孢菌素中的一种或其组合。
[0039]
本发明的优选技术方案中,所述p-gp诱导剂选自利福平、卡马西平、苯妥英、氟氯西林、茚地那韦、奈非那韦、利托那韦、沙奎那韦、维拉帕米、阿托伐他汀、卡马西平、咖啡因、长春新碱、紫杉醇中的一种或其组合。
[0040]
本发明的优选技术方案中,所述p-gp底物药物选自地高辛、布尼洛尔、他林洛尔、塞利洛尔、多柔比星、柔红霉素、洛伐他汀中的一种或其组合。
[0041]
本发明的优选技术方案中,所述非甾体抗炎药选自阿司匹林、对乙酰氨基酚、吲哚美辛、萘普生、萘普酮、双氯芬酸、布洛芬、尼美舒利、罗非昔布、塞来昔布中的一种或其组
合。
[0042]
本发明的优选技术方案中,所述质子泵抑制剂选自奥美拉唑、兰索拉唑、泮托拉唑、雷贝拉唑、艾司奥美拉唑中的一种或其组合。
[0043]
本发明的优选技术方案中,所述h2受体抑制剂选自雷尼替丁、西咪替丁、法莫替丁、尼扎替丁、罗沙替丁、乙溴替丁、米吩替丁中的一种或其组合。
[0044]
本发明的优选技术方案中,所述抗凝血药物选自普通肝素、低分子肝素(依诺肝素、达肝素)、肝素衍生物(磺达肝葵钠)、华法林、利伐沙班、阿哌沙班中的一种或其组合。
[0045]
除非另有说明,本发明涉及液体与液体之间的百分比时,所述的百分比为体积/体积百分比;本发明涉及液体与固体之间的百分比时,所述百分比为体积/重量百分比;本发明涉及固体与液体之间的百分比时,所述百分比为重量/体积百分比;其余为重量/重量百分比。
[0046]
与现有技术相比,本发明具有下述有益的技术效果:
[0047]
1、本发明的制备方法通过控制杂质a的产生进而显著减少杂质b的生成并实现了达比加群酯甲磺酸盐中杂质c的控制,有效解决杂质b结构与达比加群酯极其相近而难以有效去除而影响药品质量的技术难题,且显著提高了药品质量并保障了用药安全性。
[0048]
2、本发明的纯化方法具有操作简便,显著缩短生产周期并显著降低其成本,且具有绿色环保、能耗少、利于工业化生产等优点。
附图说明
[0049]
图1实施例8达比加群酯的纯度图谱;
[0050]
图2实施例8达比加群酯甲磺酸盐的纯度图谱。
具体实施方式
[0051]
下面列举一部分具体实施例对本发明进行说明,有必要在此指出的是以下具体实施例只用于对本发明作进一步说明,不代表对本发明保护范围的限制。其他人根据本发明做出的一些非本质的修改和调整仍属于本发明的保护范围。
[0052]
实施例1达比加群酯的制备
[0053]
(1)化合物2的合成
[0054]
控制反应瓶内温度低于25℃,向盛有盐酸乙醇溶液(其中乙醇640g,总重1040g)的反应瓶中加入对甲苯磺酸一水合物(266g)、3-[[[2-[[(4-氰基苯基)氨基]甲基]-1-甲基-1h-苯并咪唑-5-基]羰基]吡啶-2-基氨基]丙酸乙酯(化合物1)(348g),保温25-30℃,搅拌,反应20h后,生成过渡态(亚胺酯盐酸盐),将其缓慢流加至盛有浓氨水(1800g)和乙醇(640g)混合溶液的反应瓶中,控制流加时间为1-1.5h,反应温度不高于30℃。流加完毕,保温20~30℃,搅拌2小时后,加入新制的3%氢氧化钠(其中氢氧化钠75.0g,水重2400g)水溶液(2475.0g),室温搅拌1h,继续降温至0-10℃,保温搅拌2h,放料,离心至无液体滴出,纯化水洗至ph7-8,得到含化合物2的滤饼。将滤饼用乙醇(1680g)和纯化水(80g)进行回流精制,得到350g的3-[[[2-[[(4-脒基苯基)氨基]甲基]-1-甲基-1h-苯并咪唑-5-基]羰基]吡啶-2-基氨基]丙酸乙酯对甲苯磺酸盐(化合物2),其中,杂质a含量1.78%。
[0055]
(2)达比加群酯的合成
[0056]
将步骤(1)所得3-[[[2-[[(4-脒基苯基)氨基]甲基]-1-甲基-1h-苯并咪唑-5-基]羰基]吡啶-2-基氨基]丙酸乙酯对甲苯磺酸盐(化合物2)100g、二氯甲烷1300g和三乙胺75g(摩尔当量为5.0eq)加至反应釜中,搅拌,10-20℃下缓慢加入氯甲酸正己酯66g(摩尔当量为2.7eq)和二氯甲烷100g,反应完全后依次用水1000g搅洗15min,分出有机相用1.1%稀盐酸溶液1000g搅洗30min,再分出有机相用饱和碳酸氢钠溶液500g搅洗15min,最后分出有机相进行减压浓缩至干,加入乙酸乙酯530g,升温至50-55℃打浆1h,缓慢降温至0-10℃,保温搅拌1h,放料离心,滤饼用乙酸乙酯180g淋洗一次,离心至干,滤饼用乙酸乙酯淋洗,保温40-50℃搅拌1h后缓慢降温至0-10℃,保温搅拌1h,放料离心,滤饼用乙酸乙酯180g淋洗一次,离心至干,40-50℃真空干燥。制得53g达比加群酯,纯度为98.53%,其中,杂质b含量0.49%。
[0057]
实施例2化合物2的纯化
[0058]
将实施例1制备的5g 3-[[[2-[[(4-脒基苯基)氨基]甲基]-1-甲基-1h-苯并咪唑-5-基]羰基]吡啶-2-基氨基]丙酸乙酯对甲苯磺酸盐(化合物2)、25ml丙酮加入反应瓶中,升温至50℃,搅拌15min,过滤,用10ml丙酮淋洗,干燥,得到化合物2精制品,其中,杂质a含量1.37%。
[0059]
实施例3化合物2的纯化
[0060]
将实施例1制备的5g 3-[[[2-[[(4-脒基苯基)氨基]甲基]-1-甲基-1h-苯并咪唑-5-基]羰基]吡啶-2-基氨基]丙酸乙酯对甲苯磺酸盐(化合物2)、25ml四氢呋喃加入反应瓶中,升温至50℃,搅拌15min,过滤,用10ml四氢呋喃淋洗,干燥得到化合物2精制品,其中,杂质a含量1.25%。
[0061]
实施例4化合物2的纯化
[0062]
将实施例1制备的5g 3-[[[2-[[(4-脒基苯基)氨基]甲基]-1-甲基-1h-苯并咪唑-5-基]羰基]吡啶-2-基氨基]丙酸乙酯对甲苯磺酸盐(化合物2)、25ml dmf加入反应瓶中,回流,搅拌15min,降温至4℃析晶,过滤,用10ml dmf淋洗,干燥得到化合物2精制品,其中,杂质a含量16.20%。
[0063]
实施例5化合物2的纯化
[0064]
将实施例1制备的5g 3-[[[2-[[(4-脒基苯基)氨基]甲基]-1-甲基-1h-苯并咪唑-5-基]羰基]吡啶-2-基氨基]丙酸乙酯对甲苯磺酸盐(化合物2)、10ml dmso、15ml丙酮加入反应瓶中,升温至50℃,搅拌15min,过滤,用10ml丙酮淋洗,干燥得到化合物2精制品,其中,杂质a含量1.42%。
[0065]
实施例6化合物2的纯化
[0066]
将实施例1制备的5g 3-[[[2-[[(4-脒基苯基)氨基]甲基]-1-甲基-1h-苯并咪唑-5-基]羰基]吡啶-2-基氨基]丙酸乙酯对甲苯磺酸盐(化合物2)、25ml乙醇加入反应瓶中,回流,搅拌15min,降温至4℃析晶,过滤,用10ml乙醇淋洗,干燥得到化合物2精制品,其中,杂质a含量1.70%。
[0067]
实施例7化合物2的纯化
[0068]
将实施例1制备的5g 3-[[[2-[[(4-脒基苯基)氨基]甲基]-1-甲基-1h-苯并咪唑-5-基]羰基]吡啶-2-基氨基]丙酸乙酯对甲苯磺酸盐(化合物2)、5mldmf、20ml水加入反应瓶中,60℃,搅拌15min,降温至4℃析晶,过滤,用10ml水淋洗,干燥得到化合物2精制品,其中,
杂质a含量0.77%。
[0069]
实施例8
[0070]
(1)化合物2的合成
[0071]
向干燥洁净的500l反应釜中,加入225kg 30~40%hcl/乙醇,搅拌下加入24.4kg对甲苯磺酸一水合物,搅拌至溶清。控温20-30℃,将47kg化合物1分批加入,约1-2小时,加完后继续通入干燥hcl气体,通hcl气体至饱和。搅拌,至反应完全。控温30℃以下,滴加氨水调节ph值至9~10,控温至25-30℃,搅拌至反应完全,降温至5℃以下,保温养晶3-4小时,离心,滤饼用600kg纯化水室温下打浆30分钟,离心,用120kg纯化水分2次淋洗,滤饼再用150kg乙醇在室温下打浆0.5h,降温至0℃以下,析晶1小时,甩干,用40kg乙醇淋洗。滤饼在50℃鼓风烘干,得52.5kg化合物2粗品,其中,杂质a含量为0.68%。
[0072]
向1000l釜中加入480kg纯化水,升温至45-50℃,备用。另一500l釜中加入115kg dmf,加入48kg化合物2粗品,加热至60c-70℃溶解,将溶液抽至上述热水中,控温在50-55℃搅拌15min后,离心。用200kg40~50℃纯化水冲釜并淋洗滤饼,滤饼50℃鼓风烘干得39.5kg化合物2精制品,其中,杂质a含量为0.27%。
[0073]
(2)达比加群酯的合成
[0074]
向1000l反应釜中,加入82kg dmf,加入28.5kg步骤(1)制得的化合物2,搅拌下加入21.5kg三乙胺。搅拌0.5h,氮气保护,加入215kg二氯甲烷,降温至5-10℃,滴加42.4kg氯甲酸正已酯二氯甲烷溶液(由17.4kg氯甲酸正已酯溶解于25kg二氯甲烷制成),控制温度5℃-15℃滴加完毕,搅拌至反应完全,加入360kg水,分相,水相用二氯甲烷215kg x2次提取,合并有机相,用228kg 5%碳酸钾溶液洗涤,有机相用水228kg x2分两次洗涤,分相,有机相加入25kg无水硫酸镁,干燥,过滤,滤饼用20kg二氯甲烷淋洗。滤液减压浓缩,加入165kg乙酸乙酯,25℃搅拌4h,降温至5℃析晶3小时,离心,用15kg乙酸乙酯淋洗,45℃鼓风干燥,得达比加群酯23.5kg,纯度为99.47%,杂质b含量为0.02%。
[0075]
(3)达比加群酯甲磺酸盐的合成
[0076]
向50l反应釜中,加入16.5kg丙酮和2.06kg达比加群酯,搅拌升温至40℃至完全溶解,加入89.5g活性炭,保温搅拌20分钟。热过滤,滤液转入50l反应釜赵红,控制35-40℃,滴加298.7g甲磺酸的丙酮溶液。滴加完毕,保温35-40℃,反应30分钟。降温20-25℃搅拌30分钟,过滤。滤饼30-35℃真空干燥。得到浅黄色固体2.18kg,纯度为99.70%,杂质c含量为0.02%。
[0077]
实施例9化合物2的纯化
[0078]
将实施例8步骤(1)制备的4g 3-[[[2-[[(4-脒基苯基)氨基]甲基]-1-甲基-1h-苯并咪唑-5-基]羰基]吡啶-2-基氨基]丙酸乙酯对甲苯磺酸盐(化合物2)及10ml dmf加入反应瓶中,室温搅拌混合,然后加入40ml水,搅拌15min,过滤,用10ml水淋洗,干燥,得到3.37g化合物2精制品,其中,杂质a含量0.36%。
[0079]
以上对本发明具体实施方式的描述并不限制本发明,本领域技术人员可以根据本发明作出各种改变或变形,只要不脱离本发明的精神,均应属于本发明权利要求保护的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1