一种氢溴酸依他佐辛原料药杂质b及其制备方法与流程
本发明涉及但不限于药物化学领域,特别是涉及一种氢溴酸依他佐辛杂质及其制备方法。
背景技术:
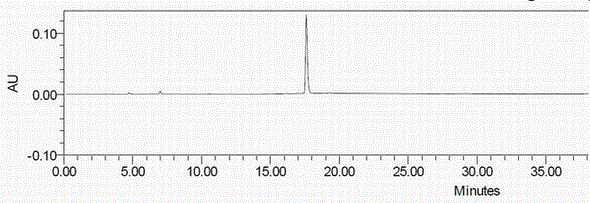
:氢溴酸依他佐辛由日本科研株式会社(nihonlyakuhinkogyoco.,ltd.)原研开发,于1987年在日本上市,主要用于治疗手术后疼痛以及癌症疼痛症,氢溴酸依他佐辛为阿片受体的部分激动剂,作用于k受体,采用选择性拮抗剂阻断突触后受体,阻断传递疼痛信息的化学信使。在镇痛方面,依他佐辛的镇痛疗效是喷他佐辛的1-2倍。氢溴酸依他佐辛结构式如下:专利ep0384917a1公开了氢溴酸依他佐辛的合成方法,该方法用化合物l-1,4-二甲基-10-甲氧基-2,3,4,5-四氢-1,6-桥亚甲基-1h-4-苯并偶氮宁-7(6h)-酮经硼氢化钠还原反应、氢化还原反应后,与氢溴酸反应脱甲基成盐得到氢溴酸依他佐辛,其反应路线为:国际人用药品注册技术协调会(ich)及欧洲药典的要求对药物中杂质需要进行严格控制,ich要求对原料药在合成,精制和储存过程中最有可能产生的实际存在的和潜在的杂质进行概述,如不对这些杂质进行严格控制,可能对人体产生严重的毒副作用。目前尚无关于氢溴酸依他佐辛原料药杂质的相关报道,为了获得高质量的氢溴酸依他佐辛原料药并进一步制备安全有效的氢溴酸依他佐辛制剂,有必要对氢溴酸依他佐辛原料药合成过程产生的杂质进行结构确证并制备出纯度较高的该杂质化合物用于氢溴酸依他佐辛原料药和制剂的质量控制过程。技术实现要素:本发明提供了一种氢溴酸依他佐辛原料药制备过程中的杂质b及其制备方法,该杂质纯度较高,可作为对照品用于氢溴酸依他佐辛原料药及制剂产品的有关物质测定及质量控制。本发明提供的一种氢溴酸依他佐辛原料药的杂质b,其特征在于,所述杂质b的结构如i式所示:该杂质是氢溴酸依他佐辛原料药制备过程中产生的杂质,在对按现有技术合成路线制备得到的依他佐辛原料药进行高效液相色谱分析时,该杂质的色谱峰较明显。发明人对氢溴酸依他佐辛合成工艺进行分析,推测产生该杂质的方式可能为中间体化合物(硼氢化钠还原产物)在氢化还原反应中未反应完全,后续在用氢溴酸脱甲基成盐反应产生:但发明人按照上述反应式制备式i化合物,反应液成分太复杂,尝试多种方法仍未能提纯出式i化合物。为了确定该杂质结构是否与式i化合物一致,必须先制备出足够纯度的式i化合物进行结构确证。发明人经过研究,找到制备出高纯度式i化合物的方法,制备出的式i化合物经高效液相色谱与氢溴酸依他佐辛原料药对比,确定为氢溴酸依他佐辛原料药中存在的杂质b。本发明提供的上述氢溴酸依他佐辛杂质b的制备方法,其特征在于,所述制备方法按以下反应式制备:具体包含以下步骤:步骤1):在惰性气体保护下,化合物ⅱ加入无水乙醇溶解,加入硼氢化钠室温搅拌反应,反应完毕将反应液减压浓缩至干;步骤2):将步骤1)所得浓缩物加入纯水溶解,用酸调节ph至7-8,减压浓缩至干,加入无水乙醇溶解,过滤除去固体,将滤液浓缩干得到化合物ⅰ粗品;步骤3):化合物ⅰ粗品用反相层析柱纯化。本发明的实施方案中,步骤1)所述惰性气体为氮气或者氩气。化合物ⅰ结构中有酚羟基,接触氧气容易氧化,因此步骤1)反应需采用惰性气体保护,优选氮气保护。本发明的实施方案中,步骤1)中无水乙醇与化合物ⅱ的重量比为100~150:1;硼氢化钠与化合物ⅱ的重量比4~6:1,硼氢化钠分批加入以使反应平缓进行,分批加入次数优选为3~6次。本发明的实施方案中,步骤2)减压浓缩除去水的温度为50~100℃,优选减压浓缩除去水的温度为60~70℃;用酸调节反应液的ph值,所述的酸选自盐酸、硫酸、氢溴酸中一种;加入无水乙醇溶解时搅拌时间为10~60分钟,直至产品充分溶解,无水乙醇与化合物ⅱ重量比为10~50:1,优选20~30:1。本发明的实施方案中,步骤3)反相层析柱的洗脱液为有机溶剂和纯水的混合溶剂,所述有机溶剂选自无水乙醇、无水甲醇、乙腈中的一种,优选无水乙醇;洗脱液无水乙醇:纯水的体积比20:80~40:60,优选30:70。本发明通过对氢溴酸依他佐辛原料药的合成工艺的分析,制备得到了氢溴酸依他佐辛原料药合成过程中产生的化合物ⅰ即杂质b。发明人无法由杂质b的产生的反应制备得到足够纯度的产品用于氢溴酸依他佐辛原料药的质量研究,而发现采用化合物ⅱ为起始原料能够制备得到纯度达到90%以上的杂质b产品,可以做为对照品用于氢溴酸依他佐辛原料药或制剂的质量控制及有关物质测定,能够对制定和提高氢溴酸依他佐辛的质量标准提供有效的数据支持,为氢溴酸依他佐辛的用药安全提供有效保障。下面结合说明书附图和具体实施方式的实施例对本发明做进一步说明。说明书附图附图1实施例2制备得到的杂质b的hplc图谱附图2实施例2制备得到的杂质b的ms图谱附图3氢溴酸依他佐辛原料药的hplc图谱具体实施方式实施例1杂质b化合物的制备向100ml三口玻璃反应瓶中依次加入化合物ⅱ0.5g、无水乙醇60ml,开启搅拌,氮气保护下分3批加入硼氢化钠3g,反应完毕,然后将反应液于旋转蒸发仪中,控制水浴60~70℃减压浓缩至干。加入适量水溶解,用盐酸溶液调节ph为7~8,减压浓缩至干。浓缩物加入适量无水乙醇溶解后,再减压浓缩至干,重复该步骤3次得到灰色的固体化合物ⅰ浓缩物,加入无水乙醇12g,搅拌20分钟,过滤,滤液浓缩干得到化合物ⅰ的粗品。将粗品用反相层析柱纯化,洗脱液为无水乙醇:纯化水(体积比30:70),收集产品溶液,减压浓缩至干。将固体转移至真空烘箱40~50℃干燥,得到0.15g灰色固体。(收率30%,hplc纯度:92.8%)。实施例2杂质b化合物的制备向1l三口玻璃反应瓶中依次加入化合物ⅱ3.2g、无水乙醇500ml,开启搅拌,氮气保护下分5批加入硼氢化钠16g,反应完毕,然后将反应液于旋转蒸发仪中,控制水浴60~70℃减压浓缩至干。加入适量水溶解,用40%氢溴酸溶液调节ph为7~8,减压浓缩至干。浓缩物加入适量无水乙醇溶解后,再减压浓缩至干,重复该步骤3次得到灰色的固体化合物ⅰ浓缩物,加入无水乙醇80g,搅拌30分钟,过滤,滤液浓缩干得到化合物ⅰ的粗品。将粗品用反相层析柱纯化,洗脱液为无水乙醇:纯化水(体积比30:70),收集产品溶液,减压浓缩至干。将固体转移至真空烘箱40~50℃干燥,得到0.72g灰色固体。收率22.5%。质谱检测ms/es+:(m+h)+248.1;ms/es-:(m-h2o)-228.2,图谱见附图2。核磁共振检测数据1hnmrδ:1.207(3h,s,-ch3),1.839~1.883(2h,m,2h),1.950~2.002(1h,t,-h),2.335~2.388(2h,m,2h),2.632(1h,m,-h),2.730(4h,m,-ch3、-h),3.220~3.246(1h,d,-h),3.298~3.348(1h,t,-h),4.643~4.651(1h,d,-h),5.550~5.565(1h,s,-oh),6.648~6.700(2h,d,2h),7.374~7.391(1h,d,-h),9.182~9.219(1h,s,-oh)。hplc纯度:93.27%。hplc图谱见附图1。hplc(高效液相色谱)检测色谱条件:hplc仪器:waterse2696高效液相色谱仪,dad检测器;色谱条件:用十八烷基硅烷键合硅胶为填充剂;以2%冰醋酸-甲醇(65:35,每1l中含有0.8g庚烷磺酸钠)为流动相a,以2%冰醋酸-甲醇(50:50,每1l中含有0.8g庚烷磺酸钠)为流动相b,按下表进行梯度洗脱,流速为0.9ml/min,检测波长为280nm。梯度如下:实施例3氢溴酸依他佐辛原料药的制备根据专利cn104356065a公开的工艺,制备氢溴酸依他佐辛,采用与实施例1相同的色谱条件对产品进行hplc检测,图谱见附图3,其中,包含氢溴酸依他佐辛及其所含杂质b的各色谱峰出峰情况如下:名称出峰时间(min)峰面积含量(%)峰高杂质b17.608127670.151348n/a19.266500600.586512氢溴酸依他佐辛21.623859211898.88682547n/a31.120341100.391804与实施例2制得化合物ⅰ的hplc图谱对照可知,化合物i与杂质b的出峰时间相同,从而可确定杂质b为化合物i的结构。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1