一种带保护基多取代含氟六元含氮杂环甲胺的制备方法与流程
本发明涉及药物中间体制备的技术领域,尤其涉及一种带保护基多取代含氟六元含氮杂环甲胺的制备方法。
背景技术:
4-羟基苯甲酸及其衍生物是一类重要的化合物,具有较强的生物活性,广泛应用于医药、农药等领域,因此4-羟基苯甲酸衍生物的合成受到了广泛关注,尤其是在医药中间体之中被广泛应用。cn110357773a中给出了一种3-氯-4-羟基苯甲酸的制备方法,cn109096099a中给出了一种3,5-二叔丁基-4-羟基苯甲酸的合成方法,这都是4-羟基苯甲酸衍生物应用和合成的示例,但是本申请提及的((2-二氟甲氧基)-5-羟基吡啶-3-基)甲基)氨基甲酸叔丁酯鲜少有作为医药中间体的应用出现,或者有文献或者资料公开出来。由于该分子的特性,独特的合成路线,独有的较高的收率问题,该方法不能推广到其他类似结构的合成中。
由于该分子的特性,该方法不能推广到其他类似结构的合成中。这是由无数次试验得到的该制备方法的独创性、较高产率和较短反应时间的不可复制性决定的,其他路线基本无法有较高产率或者能够接受的反应时间。
此外,现有技术还存在着这样的问题,即现有的萃取装置的不足之处,以本申请的反应剂量而言,远超出了一般简单定性的剂量,在这种剂量之下,简单的萃取器械不敷使用,如果使用常见的分液萃取装置,每次只能萃取最多300-400ml,一次萃取就要分开好多次,需要多次分离合并,及其费时费力,虚耗精力,对于合成人员而言,将大量时间精力用于这种环节,非常不值得。目前也有一些公司可以订制较大的萃取器材,但是只是将萃取器材做大,并不能解决问题,因为萃取器材由于溶解物性质的问题,一般必须是玻璃器材才能耐用,但是过大的玻璃器材晃动、搬动均不容易,虽然能够一次承装大量液体混合物,但是萃取操作上难以握持,晃动震动不便,容易摔坏,又给实际操作增加了难度,现有技术中并没有手段同时解决两者问题,即过小的萃取装置容积过小,以及过大的萃取装置震动操作不便的问题,提供一种可操作性强,又能一次进行大体积液体萃取的装置。
本申请的((2-二氟甲氧基)-5-羟基吡啶-3-基)甲基)氨基甲酸叔丁酯的制备中就存在这样的问题,一次萃取光溶剂就500ml,小器材装不下,分几次非常麻烦,大型器材动辄几升的玻璃器皿,拿不动,不敢晃,一次失误整个实验就报废,浪费又危险。
技术实现要素:
本发明的第一目的是解决现有技术中两个具体问题,一是从2,3-二氟苯胺出发,如何经过不多且产率又高的步骤得到((2-二氟甲氧基)-5-羟基吡啶-3-基)甲基)氨基甲酸叔丁酯的问题,本申请方案完美解决了该问题,二是面对萃取环节实施难度大,过小的萃取装置容积过小,以及过大的萃取装置震动操作不便的问题,这在本申请中都得到了解决。
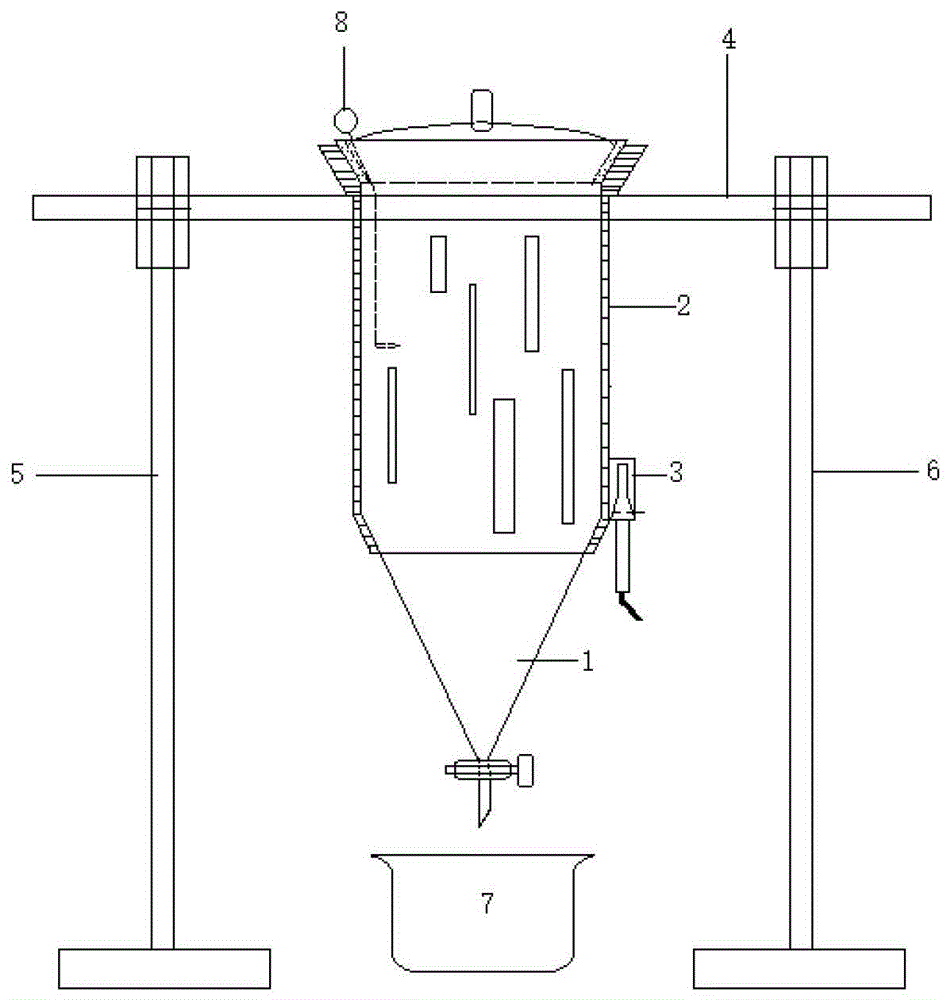
本发明要求保护一种超声萃取装置,其特征在于:包括萃取部1、外壳部2、超声部3、横杆部4、左支撑部5、右支撑部6、玻璃器7、破乳组件8。
所述萃取部1包括圆柱部11、上开口12、上盖13、锥体部14、止液栓组15、出液口16。所述萃取部1的主体部分自上而下地由上开口12、圆柱部11、锥体部14拼接而成,圆柱部11呈空心圆柱型,上开口12上大下小且内侧面为磨砂部121,上盖13与磨砂部121适配的外缘为磨砂面,上盖13上方中央具有手柄部131,锥体部14呈中空的圆锥型,锥体部14和出液口16之间具有止液栓组15,止液栓组15具有一止液腔153,止液腔153内具有一个与液体通路相垂直的水平通孔,其中塞入止液栓151,止液栓一侧具有止液栓柄152。
所述外壳部2包括下凹圈21、主壳22、观察槽23、周缘24、承托部25和振头槽27,所述下凹圈21与椎体部14的最上部贴合,所述主壳22和圆柱部11贴合,所述所述观察槽23为竖直地开在主壳(22)上的若干通槽,该若干通槽为高宽不等位置不定的若干矩形通槽,所述周缘24为承托部25下部的一周凸出边缘,所述承托部25内侧承托所述上开口12。所述主壳22上具有一个与振头槽27左侧相匹配的凹坑,所述振头槽27具有一个向下的与振头31适配的凹槽。
所述超声装置3包括振头31、换能器32、发生器33、电源34、加强电缆35,振头31和换能器32通过加强电缆35连接,振头31和换能器32的连接线包裹在加强电缆35之内,换能器32和发生器33连接,发生器33由电源34供电。所述振头31插入凹槽中,并由凹槽螺丝36固定。
所述横杆部4中间为圆环部42,其内径与主壳22外径适配,周缘24得以被圆环部42上部承托,圆环部42左右分别与圆柱形的左横杆41和右横杆43一体连接。
所述左支撑部5包括左杆51、左底座52、左上环53、左铰接部54、左下环55、左凸缘56、左轴承57,左支撑部5位于萃取部1左方,左底座52为中间具有一螺孔的圆柱形底座,左杆51下端向该螺孔内旋入固定,左杆51上端焊接连接左下环55,左下环55与上方同为半环形的左上环53扣合,左下环55和左上环53左侧通过左铰接部54铰接,右侧具有一对相适配的左凸缘56,所述左凸缘56由螺栓组固定,扣合的左下环55和左上环53内固定有左轴承57,左轴承57内孔插入左横杆41。
所述右支撑部6包括右杆61、右底座62、右上环63、右铰接部64、右下环65、右凸缘66、右轴承57,右支撑部6位于萃取部1右方,右底座52为中间具有一螺孔的圆柱形底座,右杆61下端向该螺孔内旋入固定,右杆61上端焊接连接右下环65,右下环65与上方同为半环形的右上环63扣合,右下环65和右上环63右侧通过右铰接部64铰接,左侧具有一对相适配的右凸缘66,所述右凸缘56由螺栓组固定,扣合的右下环65和右上环63内固定有右轴承67,右轴承67内孔插入右横杆43。
所述玻璃器7位于出液口16下方。
所述破乳组件8包括球部81、片部82和锥部83。
优选地,所述萃取部材质为玻璃。所述水平通孔内涂有凡士林。
所述外壳部为尼龙pa66、聚四氟乙烯或工程塑料材质的任一种。
所述振头槽27是使用电工胶布多圈缠绕固定在主壳22上。
所述振头的工作功率在60-100w之间,工作频率在15khz-100khz之间,振头头部为喇叭形附带一伸出的直柱。
所述横杆部4、左支撑部5、右支撑部6的材质为不锈钢。
所述左轴承和右轴承均是滚珠轴承。所述左轴承和右轴承内外均可配设橡胶垫圈。
所述玻璃器7是大容积广口容器或大口径漏斗与其他玻璃器皿的组合。
所述破乳组件8是聚四氟乙烯材质。
一种带保护基多取代含氟六元含氮杂环甲胺的制备方法,由于其需要的萃取剂量大,因此使用如前所述的一种超声萃取装置以进行,其特征在于,包括以下步骤。
(1)将9-11g的2-羟基-5-溴吡啶-3-甲酸溶于105-115ml甲醇中,搅拌至体系呈无色透明,升温至65℃回流,开始滴加13-14g氯化亚砜,6-8min滴加完毕,体系逐渐变为黄色透明,65℃保温直至通过hplc监测反应完毕,再将反应混合物降至室温,将反应混合物旋干浓缩,加入50ml乙酸乙酯稀释,再加入20ml水洗涤,析出固体,将反应混合物全部抽滤,固体用50ml甲基叔丁基醚打浆再抽滤,烘干后得2-羟基-5-溴吡啶-3-甲酸甲酯。
(2)将10-11g的2-羟基-5-溴吡啶-3-甲酸甲酯溶于550ml乙腈中,用外置的冰水浴将体系降温至0℃,将体系保持搅拌并保持外置冰水浴,缓慢加入4.8-5.2g的60%的氢化钠,加入完毕后滴加3.85-4.15g的2-氟磺酰基二氟乙酸,用至少10min滴加完毕,撤掉冰水浴缓慢升至常温,持续反应直至用hplc与lcms监测反应完毕后,滴加20ml饱和nh4cl溶液与30mlh2o淬灭氢化钠,溶液逐渐变为澄清的黑褐色液体并析出固体,全部抽滤,再向混合液中加入600ml乙酸乙酯进行萃取并分离有机相,再用每次100ml饱和氯化钠溶液洗涤有机相2次,有机相干燥旋干,5倍拌样质量拌样,石油醚:乙酸乙酯=7:1的前提下200-300硅胶过柱,得2-二氟甲氧基-5-溴吡啶-3-甲酸甲酯。
(3)将9.5-10.5g的2-二氟甲氧基-5-溴吡啶-3-甲酸甲酯溶于50ml水与50ml四氢呋喃的混合液中,用外置冰水浴将体系降温至约10℃,向体系中缓慢加入4.25-4.75g的一水合氢氧化锂,至少用2-4min加入完毕,反应至少2.5-3.5h后,通过hplc与lcms监测反应完毕后,向体系中滴加12n的hcl直至将ph调至约2,然后每次加入100ml二氯甲烷对混合物进行萃取2次,合并有机相干燥旋干,得2-二氟甲氧基-5-溴吡啶-3-甲酸。
(4)将8-9g的2-二氟甲氧基-5-溴吡啶-3-甲酸全部加入到80ml三氯甲烷中,用外置冰水浴将体系降温至0-5℃,然后滴入15-17ml氯化亚砜,滴完后将体系升温至50℃反应30分钟,将反应混合物全部旋干,将残余物溶于50ml四氢呋喃,然后滴入100ml预冷至0-10℃之间的氨水,将体系自然升至室温,反应8-16h,旋掉全部溶剂,析出固体,抽滤,用50ml水淋洗,烘干,得到2-二氟甲氧基-5-溴吡啶-3-甲酰胺。
(5)将7-7.5g的2-二氟甲氧基-5-溴吡啶-3-甲酰胺加入到100ml二氯甲烷中,然后加入20-22ml三乙胺,用外置冰水浴将反应混合物降温至0-5℃,滴入13-15g三氟乙酸酐,滴完后将反应混合物自然升至室温,反应16小时,倒入100ml冰水混合物中,分液,水相用50ml二氯甲烷萃取一次,然后有机相再用50ml饱和食盐水洗一次,无水硫酸钠干燥,抽滤,旋干得2-二氟甲氧基-3-氰基-5-溴吡啶。
(6)在氮气保护下,将2.2-2.7g的2-二氟甲氧基-3-氰基-5-溴吡啶溶于30ml四氢呋喃中,用外置冰水浴将反应混合物降温至0℃,滴加入40ml硼烷四氢呋喃溶液,滴加完毕后自然升至室温,保温反应1h,再升温至出现回流,保持反应过夜,中途降温取样,将取出的样品慢慢滴加到甲醇和水中,送hplc-lcms监测反应完毕后,将反应混合物降至0℃,慢慢滴加入70ml甲醇,滴加完毕后,慢慢升至室温,搅拌至少1-2h,然后回流至少1-2h,除去溶剂得到油状液体2-二氟甲氧基-5-溴吡啶-3-甲胺。
(7)将步骤6得到的全部2-二氟甲氧基-5-溴吡啶-3-甲胺溶于30ml甲基叔丁基醚,将混合物用冰盐浴降温至-10℃,滴加入至少2.5eq的7n的二氧六环/hcl,搅拌反应至少1-2h,抽滤得到2-二氟甲氧基-5-溴吡啶-3-甲胺盐酸盐固体。
(8)将步骤7得到的2-二氟甲氧基-5-溴吡啶-3-甲胺盐酸盐在20ml二氯甲烷内充分分散,在低速搅拌下加入2.7-3.3g三乙胺,保持常温下分批加入3-3.6g的boc酸酐,反应3h,加入10ml水,10mlx3二氯甲烷分液,盐水洗,干燥,除去溶剂,柱分离得到boc保护2-二氟甲氧基-5-溴吡啶-3-甲胺。
(9)在搅拌下,将3.8-4.2g的boc保护2-二氟甲氧基-5-溴吡啶-3-甲胺溶于120ml二氧六环中,在室温下将5.5-6.1g的双戊酰二硼与3-3.5g的乙酸钾缓慢加入至体系中,保持搅拌时体系充分分散,将体系抽冲氮气三次后,在氮气保护下向体系中加入450-500mg的pd(dppf)cl2-ch2cl2,升温至85℃回流后保持温度,反应至少10-14h,hplc监测反应完毕后,冷却至室温,加入360ml乙酸乙酯稀释,抽滤后旋干拌样,石油醚:乙酸乙酯=20:1条件下,5倍拌样200-300硅胶过柱,得到产品,用30ml石油醚打浆30min,抽滤,得到纯品((2-二氟甲氧基-5-频那醇酯基)吡啶-3-基)甲基)氨基甲酸叔丁酯。
(10)将20-22g的nabo3-4h2o溶于40mlh2o中,用外置冰水浴降温至0℃,将18-20g((2-二氟甲氧基-5-频那醇酯基)吡啶-3-基)甲基)氨基甲酸叔丁酯溶于360ml四氢呋喃后滴加进体系,滴加完毕后升至室温,至少反应2-4h后,向体系中加入400ml饱和氯化铵与400ml乙酸乙酯,分液后,水相用400ml乙酸乙酯再次萃取,有机相用400mlh2o洗涤后,干燥,旋干拌样,石油醚:乙酸乙酯=10:1条件下,5倍拌样200-300硅胶过柱,得产物((2-(二氟甲氧基)-5-羟基吡啶-3-基)甲基)氨基甲酸叔丁酯。
前述(1)-(10)中的萃取,利用如权利要求2所述的超声萃取装置实施,具体步骤为。
(a)准备步骤:清洗并晾干萃取部,将萃取部完全套入外壳部内固定,再将外壳部套入圆环部内,以使得周缘(24)得以被圆环部(42)上部承托,将振头槽(27)左侧顶入主壳(22)上的凹坑内,使用电工胶布在振头槽高摄氏度多圈缠绕,将其固定在主壳(22)上。
(b)盖上步骤:打开上盖(13),将欲萃取的混合物和溶剂倒入,放置破乳组件使球部在上开口之外且锥部向内,小心盖上上盖。
(c)震动晃动步骤:推动外壳部,使外壳部钟摆式前后晃动10-20次,手动空直外壳部大致静止,启动超声装置并使得振头以20khz以上的频率震动1-2min,静置2-4min。
(d)重复步骤:重复步骤c3-5次。
(e)接取步骤:停止萃取,打开上盖,拿出破乳组件,用不同的玻璃器分别在出液口下方接取不同相态的混合物,萃取部的最后一种相态可从上口倒出。
优选地,前述所有试剂均为化学纯以上纯度,或者均为优级纯。所述水均为去离子水,优选地为双蒸水。
与现有技术相比,本发明的优点在于:一、在装置方面改进,该装置完美滴解决了两个问题;面对萃取环节实施难度大,过小的萃取装置容积过小,以及过大的萃取装置震动操作不便的问题;再者,本申请的超声装置结合方式独具特色,如果事先固定,则无法放入,而这种凹槽加胶带的方式,固定强度够,又易于拆卸,外壳2的形状解决了大型玻璃器的固定问题。观察槽解决了固定后的液面观察问题,可以透过其观察有机相的分离程度,非常方便,另外越是体积大,越存在破乳难题,聚四氟做的破乳组件用整块聚四氟削成,可以有效地解决破乳问题。还有就是,在下凹圈承托压力大时,可以为其外设有弧度的金属圈辅助固定,不用时可拆下。
目前现有技术中并无该产物制得的报道,与同类物质制备方法相比,本申请方法步骤精细考究,每一步原料利用率均非常高,有极大价值实现工业生产,通过本发明的方法精细设计,不仅有效地实现了合成,且产率较高,2-羟基-5-溴吡啶-3-甲酸(10g)可以获得1克((2-二氟甲氧基)-5-羟基吡啶-3-基)甲基)氨基甲酸叔丁酯,有一定的工业生产价值,经济价值较大,本申请通过对于萃取环节的精心设计,体现了极强的发明构思和创造性,取得了良好的制备效果,在现有技术中并无较为类似的公开信息可供借鉴,本发明方案具有独创性。与之相对的是,利用传统萃取方式进行制备,在萃取环节至少要浪费一倍的时间,可以说没有实用价值。传统萃取设备容积不足,难以进行如上大量溶剂下的萃取,而且转移物料困难,在每次转移出和混合、分离时都要损失大部分,在物料少时,大量物质多次粘附在玻璃器的各个口颈处,进而严重影响了产率,申请人将萃取环节用传统器具进行,时间延长了一倍有余,相较本申请方法,产率至少降低了30%。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1是装置整体示意图。
图2是左支撑部、右支撑部的侧向结构示意图。
图3是本发明的横杆部俯视示意图。
图4是本发明的超声装置视图。
图5是萃取部组成示意图。
图6是外壳部组成示意图。
图7是破乳组件侧向和正向示意图。
图8是((2-二氟甲氧基)-5-羟基吡啶-3-基)甲基)氨基甲酸叔丁酯的核磁图。
图9是((2-二氟甲氧基)-5-羟基吡啶-3-基)甲基)氨基甲酸叔丁酯的合成路线图。
附图标记:萃取部1、圆柱部11、上开口12、磨砂部121、上盖13、手柄部131、锥体部14、止液栓组15、止液栓151、止液栓柄152、止液腔153、出液口16、外壳部2、下凹圈21、主壳22、观察槽23、周缘24、承托部25、振头槽27、超声部3、振头31、换能器32、发生器33、电源34、加强电缆35、凹槽螺丝36、横杆部4、左横杆41、圆环部42、右横杆43、左支撑部5、左杆51、左底座52、左上环53、左铰接部54、左下环55、左凸缘56、左轴承57、右支撑部6、右杆61、右底座62、右上环63、右铰接部64、右下环65、右凸缘66、右轴承57、玻璃器7、破乳组件8、球部81、片部82、锥部83。
具体实施方式
下面结合附图对本发明的优选实施例进行详细阐述,以使本发明的优点和特征能更易于被本领域技术人员理解,从而对本发明的保护范围做出更为清楚明确的界定。
实施例1
一种超声萃取装置,其特征在于:包括萃取部1、外壳部2、超声部3、横杆部4、左支撑部5、右支撑部6、玻璃器7、破乳组件8。所述萃取装置可分为多个规格,例如萃取部的容积可以为1l、2l、3l、4l、5l,以应对不同需求。
所述萃取部1包括圆柱部11、上开口12、上盖13、锥体部14、止液栓组15、出液口16。所述萃取部1的主体部分自上而下地由上开口12、圆柱部11、锥体部14拼接而成,圆柱部11呈空心圆柱型,上开口12上大下小且内侧面为磨砂部121,上盖13与磨砂部121适配的外缘为磨砂面,上盖13上方中央具有手柄部131,锥体部14呈中空的圆锥型,锥体部14和出液口16之间具有止液栓组15,止液栓组15具有一止液腔153,止液腔153内具有一个与液体通路相垂直的水平通孔,其中塞入止液栓151,止液栓一侧具有止液栓柄152。为了防止液体压力过高导致止液栓被顶出,止液栓可配有皮筋或者专用的橡胶固定器,在加入液体之前就放置,用于固定止液栓柄152,防止被过大的压力顶出。
所述外壳部2包括下凹圈21、主壳22、观察槽23、周缘24、承托部25和振头槽27,所述下凹圈21与椎体部14的最上部贴合,所述主壳22和圆柱部11贴合,所述所述观察槽23为竖直地开在主壳(22)上的若干通槽,该若干通槽为高宽不等位置不定的若干矩形通槽,所述周缘24为承托部25下部的一周凸出边缘,所述承托部25内侧承托所述上开口12。所述主壳22上具有一个与振头槽27左侧相匹配的凹坑,所述振头槽27具有一个向下的与振头31适配的凹槽。下凹圈21、主壳22、观察槽23、周缘24、承托部25一体成型,对于较重的情形,为了防止下凹圈21变形导致萃取部滑出摔碎,可以在下凹圈21外部配有金属固定带,可以是铝合金制,用法兰和螺栓固定紧,其弧度紧贴下凹圈外部。
所述超声装置3包括振头31、换能器32、发生器33、电源34、加强电缆35,振头31和换能器32通过加强电缆35连接,振头31和换能器32的连接线包裹在加强电缆35之内,换能器32和发生器33连接,发生器33由电源34供电。所述振头31插入凹槽中,并由凹槽螺丝36固定。所述振头的功率在60-100w之间,频率在15khz-100khz之间的某整数,振头某段为喇叭形附带一伸出的直柱,凹槽设置为与其同型适配,且凹槽上还具有对称的至少两个通孔,用于凹槽螺丝36透过且穿入振头上的螺丝孔而固定。
所述横杆部4中间为圆环部42,其内径与主壳22外径适配,周缘24得以被圆环部42上部承托,圆环部42左右分别与圆柱形的左横杆41和右横杆43一体连接。圆环部42与左横杆41和右横杆43可以是一体成型的不锈钢,也可以是焊接而成。
所述左支撑部5包括左杆51、左底座52、左上环53、左铰接部54、左下环55、左凸缘56、左轴承57,左支撑部5位于萃取部1左方,左底座52为中间具有一螺孔的圆柱形底座,左杆51下端向该螺孔内旋入固定,左杆51上端焊接连接左下环55,左下环55与上方同为半环形的左上环53扣合,左下环55和左上环53左侧通过左铰接部54铰接,右侧具有一对相适配的左凸缘56,所述左凸缘56由螺栓组固定,扣合的左下环55和左上环53内固定有左轴承57,左轴承57内孔插入左横杆41。所述左轴承是滚珠轴承,内圈和外圈之间有一圈滚珠。
所述右支撑部6包括右杆61、右底座62、右上环63、右铰接部64、右下环65、右凸缘66、右轴承57,右支撑部6位于萃取部1右方,右底座52为中间具有一螺孔的圆柱形底座,右杆61下端向该螺孔内旋入固定,右杆61上端焊接连接右下环65,右下环65与上方同为半环形的右上环63扣合,右下环65和右上环63右侧通过右铰接部64铰接,左侧具有一对相适配的右凸缘66,所述右凸缘56由螺栓组固定,扣合的右下环65和右上环63内固定有右轴承67,右轴承67内孔插入右横杆43。所述左轴承是滚珠轴承,内圈和外圈之间有一圈滚珠。
所述玻璃器7位于出液口16下方。所述破乳组件8包括球部81、片部82和锥部83。聚四氟做的破乳组件用整块聚四氟削成,球部用于坠住整个组件,片部非常薄,锥部非常细,尖端为针状。破乳组件在实践中可以布置多个锥部高低不同的多个。
优选地,所述萃取部材质为玻璃。所述水平通孔内涂有凡士林。所述外壳部为尼龙pa66、聚四氟乙烯或工程塑料材质的任一种。所述振头槽27是使用电工胶布多圈缠绕固定在主壳22上。所述振头的工作功率在60-100w之间,工作频率在15khz-100khz之间,振头头部为喇叭形附带一伸出的直柱。所述横杆部4、左支撑部5、右支撑部6的材质为不锈钢。所述左轴承和右轴承均是滚珠轴承。所述左轴承和右轴承内外均可配设橡胶垫圈。所述玻璃器7是大容积广口容器或大口径漏斗与其他玻璃器皿的组合,例如是大烧杯、或广口瓶。所述破乳组件8是聚四氟乙烯材质。
实施例2
一种带保护基多取代含氟六元含氮杂环甲胺的制备方法,由于其需要的萃取剂量大,因此使用如前所述的一种超声萃取装置以进行,其特征在于,包括以下步骤。
(1)将9-11g的2-羟基-5-溴吡啶-3-甲酸溶于105-115ml甲醇中,搅拌至体系呈无色透明,升温至65℃回流,开始滴加13-14g氯化亚砜,6-8min滴加完毕,体系逐渐变为黄色透明,65℃保温直至通过hplc监测反应完毕,再将反应混合物降至室温,将反应混合物旋干浓缩,加入50ml乙酸乙酯稀释,再加入20ml水洗涤,析出固体,将反应混合物全部抽滤,固体用50ml甲基叔丁基醚打浆再抽滤,烘干后得2-羟基-5-溴吡啶-3-甲酸甲酯。
(2)将10-11g的2-羟基-5-溴吡啶-3-甲酸甲酯溶于550ml乙腈中,用外置的冰水浴将体系降温至0℃,将体系保持搅拌并保持外置冰水浴,缓慢加入4.8-5.2g的60%的氢化钠,加入完毕后滴加3.85-4.15g的2-氟磺酰基二氟乙酸,用至少10min滴加完毕,撤掉冰水浴缓慢升至常温,持续反应直至用hplc与lcms监测反应完毕后,滴加20ml饱和nh4cl溶液与30mlh2o淬灭氢化钠,溶液逐渐变为澄清的黑褐色液体并析出固体,全部抽滤,再向混合液中加入600ml乙酸乙酯进行萃取并分离有机相,再用每次100ml饱和氯化钠溶液洗涤有机相2次,有机相干燥旋干,5倍拌样质量拌样,石油醚:乙酸乙酯=7:1的前提下200-300硅胶过柱,得2-二氟甲氧基-5-溴吡啶-3-甲酸甲酯。
(3)将9.5-10.5g的2-二氟甲氧基-5-溴吡啶-3-甲酸甲酯溶于50ml水与50ml四氢呋喃的混合液中,用外置冰水浴将体系降温至约10℃,向体系中缓慢加入4.25-4.75g的一水合氢氧化锂,至少用2-4min加入完毕,反应至少2.5-3.5h后,通过hplc与lcms监测反应完毕后,向体系中滴加12n的hcl直至将ph调至约2,然后每次加入100ml二氯甲烷对混合物进行萃取2次,合并有机相干燥旋干,得2-二氟甲氧基-5-溴吡啶-3-甲酸。
(4)将8-9g的2-二氟甲氧基-5-溴吡啶-3-甲酸全部加入到80ml三氯甲烷中,用外置冰水浴将体系降温至0-5℃,然后滴入15-17ml氯化亚砜,滴完后将体系升温至50℃反应30分钟,将反应混合物全部旋干,将残余物溶于50ml四氢呋喃,然后滴入100ml预冷至0-10℃之间的氨水,将体系自然升至室温,反应8-16h,旋掉全部溶剂,析出固体,抽滤,用50ml水淋洗,烘干,得到2-二氟甲氧基-5-溴吡啶-3-甲酰胺。
(5)将7-7.5g的2-二氟甲氧基-5-溴吡啶-3-甲酰胺加入到100ml二氯甲烷中,然后加入20-22ml三乙胺,用外置冰水浴将反应混合物降温至0-5℃,滴入13-15g三氟乙酸酐,滴完后将反应混合物自然升至室温,反应16小时,倒入100ml冰水混合物中,分液,水相用50ml二氯甲烷萃取一次,然后有机相再用50ml饱和食盐水洗一次,无水硫酸钠干燥,抽滤,旋干得2-二氟甲氧基-3-氰基-5-溴吡啶。
(6)在氮气保护下,将2.2-2.7g的2-二氟甲氧基-3-氰基-5-溴吡啶溶于30ml四氢呋喃中,用外置冰水浴将反应混合物降温至0℃,滴加入40ml硼烷四氢呋喃溶液,滴加完毕后自然升至室温,保温反应1h,再升温至出现回流,保持反应过夜,中途降温取样,将取出的样品慢慢滴加到甲醇和水中,送hplc-lcms监测反应完毕后,将反应混合物降至0℃,慢慢滴加入70ml甲醇,滴加完毕后,慢慢升至室温,搅拌至少1-2h,然后回流至少1-2h,除去溶剂得到油状液体2-二氟甲氧基-5-溴吡啶-3-甲胺。
(7)将步骤6得到的全部2-二氟甲氧基-5-溴吡啶-3-甲胺溶于30ml甲基叔丁基醚,将混合物用冰盐浴降温至-10℃,滴加入至少2.5eq的7n的二氧六环/hcl,搅拌反应至少1-2h,抽滤得到2-二氟甲氧基-5-溴吡啶-3-甲胺盐酸盐固体。
(8)将步骤7得到的2-二氟甲氧基-5-溴吡啶-3-甲胺盐酸盐在20ml二氯甲烷内充分分散,在低速搅拌下加入2.7-3.3g三乙胺,保持常温下分批加入3-3.6g的boc酸酐,反应3h,加入10ml水,10mlx3二氯甲烷分液,盐水洗,干燥,除去溶剂,柱分离得到boc保护2-二氟甲氧基-5-溴吡啶-3-甲胺。
(9)在搅拌下,将3.8-4.2g的boc保护2-二氟甲氧基-5-溴吡啶-3-甲胺溶于120ml二氧六环中,在室温下将5.5-6.1g的双戊酰二硼与3-3.5g的乙酸钾缓慢加入至体系中,保持搅拌时体系充分分散,将体系抽冲氮气三次后,在氮气保护下向体系中加入450-500mg的pd(dppf)cl2-ch2cl2,升温至85℃回流后保持温度,反应至少10-14h,hplc监测反应完毕后,冷却至室温,加入360ml乙酸乙酯稀释,抽滤后旋干拌样,石油醚:乙酸乙酯=20:1条件下,5倍拌样200-300硅胶过柱,得到产品,用30ml石油醚打浆30min,抽滤,得到纯品((2-二氟甲氧基-5-频那醇酯基)吡啶-3-基)甲基)氨基甲酸叔丁酯。
(10)将20-22g的nabo3-4h2o溶于40mlh2o中,用外置冰水浴降温至0℃,将18-20g((2-二氟甲氧基-5-频那醇酯基)吡啶-3-基)甲基)氨基甲酸叔丁酯溶于360ml四氢呋喃后滴加进体系,滴加完毕后升至室温,至少反应2-4h后,向体系中加入400ml饱和氯化铵与400ml乙酸乙酯,分液后,水相用400ml乙酸乙酯再次萃取,有机相用400mlh2o洗涤后,干燥,旋干拌样,石油醚:乙酸乙酯=10:1条件下,5倍拌样200-300硅胶过柱,得产物((2-(二氟甲氧基)-5-羟基吡啶-3-基)甲基)氨基甲酸叔丁酯。
实施例3
一种带保护基多取代含氟六元含氮杂环甲胺的制备方法,由于其需要的萃取剂量大,因此使用如前所述的一种超声萃取装置以进行,其特征在于,包括以下步骤。
(1)将11g的2-羟基-5-溴吡啶-3-甲酸溶于115ml甲醇中,搅拌至体系呈无色透明,升温至65℃回流,开始滴加14g氯化亚砜,8min滴加完毕,体系逐渐变为黄色透明,65℃保温直至通过hplc监测反应完毕,再将反应混合物降至室温,将反应混合物旋干浓缩,加入50ml乙酸乙酯稀释,再加入20ml水洗涤,析出固体,将反应混合物全部抽滤,固体用50ml甲基叔丁基醚打浆再抽滤,烘干后得2-羟基-5-溴吡啶-3-甲酸甲酯,收率为76.3%。
(2)将11g的2-羟基-5-溴吡啶-3-甲酸甲酯溶于550ml乙腈中,用外置的冰水浴将体系降温至0℃,将体系保持搅拌并保持外置冰水浴,缓慢加入5.2g的60%的氢化钠,加入完毕后滴加4.15g的2-氟磺酰基二氟乙酸,用至少10min滴加完毕,撤掉冰水浴缓慢升至常温,持续反应直至用hplc与lcms监测反应完毕后,滴加20ml饱和nh4cl溶液与30mlh2o淬灭氢化钠,溶液逐渐变为澄清的黑褐色液体并析出固体,全部抽滤,再向混合液中加入600ml乙酸乙酯进行萃取并分离有机相,再用每次100ml饱和氯化钠溶液洗涤有机相2次,有机相干燥旋干,5倍拌样质量拌样,石油醚:乙酸乙酯=7:1的前提下200-300硅胶过柱,得2-二氟甲氧基-5-溴吡啶-3-甲酸甲酯,收率为73.4%。
(3)将10.5g的2-二氟甲氧基-5-溴吡啶-3-甲酸甲酯溶于50ml水与50ml四氢呋喃的混合液中,用外置冰水浴将体系降温至约10℃,向体系中缓慢加入4.75g的一水合氢氧化锂,至少用3min加入完毕,反应至少3h后,通过hplc与lcms监测反应完毕后,向体系中滴加12n的hcl直至将ph调至约2,然后每次加入100ml二氯甲烷对混合物进行萃取2次,合并有机相干燥旋干,得2-二氟甲氧基-5-溴吡啶-3-甲酸,收率为85.8%。
(4)将9g的2-二氟甲氧基-5-溴吡啶-3-甲酸全部加入到80ml三氯甲烷中,用外置冰水浴将体系降温至0-5℃,然后滴入17ml氯化亚砜,滴完后将体系升温至50℃反应30分钟,将反应混合物全部旋干,将残余物溶于50ml四氢呋喃,然后滴入100ml预冷至0-10℃之间的氨水,将体系自然升至室温,反应14h,旋掉全部溶剂,析出固体,抽滤,用50ml水淋洗,烘干,得到2-二氟甲氧基-5-溴吡啶-3-甲酰胺,收率85.1%。
(5)将7.5g的2-二氟甲氧基-5-溴吡啶-3-甲酰胺加入到100ml二氯甲烷中,然后加入22ml三乙胺,用外置冰水浴将反应混合物降温至0-5℃,滴入15g三氟乙酸酐,滴完后将反应混合物自然升至室温,反应16小时,倒入100ml冰水混合物中,分液,水相用50ml二氯甲烷萃取一次,然后有机相再用50ml饱和食盐水洗一次,无水硫酸钠干燥,抽滤,旋干得2-二氟甲氧基-3-氰基-5-溴吡啶,收率51%。
(6)在氮气保护下,将2.7g的2-二氟甲氧基-3-氰基-5-溴吡啶溶于30ml四氢呋喃中,用外置冰水浴将反应混合物降温至0℃,滴加入40ml硼烷四氢呋喃溶液,滴加完毕后自然升至室温,保温反应1h,再升温至出现回流,保持反应过夜,中途降温取样,将取出的样品慢慢滴加到甲醇和水中,送hplc-lcms监测反应完毕后,将反应混合物降至0℃,慢慢滴加入70ml甲醇,滴加完毕后,慢慢升至室温,搅拌至少1.5h,然后回流至少1.5h,除去溶剂得到油状液体2-二氟甲氧基-5-溴吡啶-3-甲胺。
(7)将步骤6得到的的2-二氟甲氧基-5-溴吡啶-3-甲胺溶于30ml甲基叔丁基醚,将混合物用冰盐浴降温至-10℃,滴加入2.5eq的7n的二氧六环/hcl,搅拌反应至少1.5h,抽滤得到2-二氟甲氧基-5-溴吡啶-3-甲胺盐酸盐固体。
(8)将步骤7得到的2-二氟甲氧基-5-溴吡啶-3-甲胺盐酸盐在20ml二氯甲烷内充分分散,在低速搅拌下加入3.3g三乙胺,保持常温下分批加入3.6g的boc酸酐,反应3h,加入10ml水,10mlx3二氯甲烷分液,盐水洗,干燥,除去溶剂,柱分离得到boc保护2-二氟甲氧基-5-溴吡啶-3-甲胺,(6)-(8)三步总收率为27.3%。
(9)在搅拌下,将4.2g的boc保护2-二氟甲氧基-5-溴吡啶-3-甲胺溶于120ml二氧六环中,在室温下将6.05g的双戊酰二硼与3.5g的乙酸钾缓慢加入至体系中,保持搅拌时体系充分分散,将体系抽冲氮气三次后,在氮气保护下向体系中加入487mg的pd(dppf)cl2-ch2cl2,升温至85℃回流后保持温度,反应至少14h,hplc监测反应完毕后,冷却至室温,加入360ml乙酸乙酯稀释,抽滤后旋干拌样,石油醚:乙酸乙酯=20:1条件下,5倍拌样200-300硅胶过柱,得到产品,用30ml石油醚打浆30min,抽滤,得到纯品((2-二氟甲氧基-5-频那醇酯基)吡啶-3-基)甲基)氨基甲酸叔丁酯,收率为55.8%,为白色固体。
(10)将22g的nabo3-4h2o溶于40mlh2o中,用外置冰水浴降温至0℃,将19g((2-二氟甲氧基-5-频那醇酯基)吡啶-3-基)甲基)氨基甲酸叔丁酯溶于360ml四氢呋喃后滴加进体系,滴加完毕后升至室温,至少反应3h后,向体系中加入400ml饱和氯化铵与400ml乙酸乙酯,分液后,水相用400ml乙酸乙酯再次萃取,有机相用400mlh2o洗涤后,干燥,旋干拌样,石油醚:乙酸乙酯=10:1条件下,5倍拌样200-300硅胶过柱,得产物((2-(二氟甲氧基)-5-羟基吡啶-3-基)甲基)氨基甲酸叔丁酯10.583g,收率为76.9%。
经计算,总收率为2.44%,我们用同等条件,只是萃取使用普通器皿进行萃取,其他条件完全一样,结果收率只有1.93%,明显较低,而且时间要长一半左右。
实施例4
一种带保护基多取代含氟六元含氮杂环甲胺的制备方法,由于其需要的萃取剂量大,因此使用如前所述的一种超声萃取装置以进行,其特征在于,包括以下步骤。
(1)将10.0g的2-羟基-5-溴吡啶-3-甲酸(45.9mmol)溶于110ml甲醇中,搅拌至体系呈无色透明,升温至65℃回流,开始滴加13.7g氯化亚砜(114.8mmol,2.5eq),7min滴加完毕,体系逐渐变为黄色透明,65℃保温直至通过hplc监测反应完毕,再将反应混合物降至室温,将反应混合物旋干浓缩,加入50ml乙酸乙酯稀释,再加入20ml水洗涤,析出固体,将反应混合物全部抽滤,固体用50ml甲基叔丁基醚打浆再抽滤,烘干后得2-羟基-5-溴吡啶-3-甲酸甲酯8.0g,收率为75.2%,为淡褐色固体。
检测数据为,1hnmr(400hz,cdcl3):δppm11.49(brs,1h),8.30-8.34(m,2h),lcms:(retentiontime:2.407min):233.0[m+1]。
(2)将10.5g的2-羟基-5-溴吡啶-3-甲酸甲酯(45.3mmol)溶于550ml乙腈中,用外置的冰水浴将体系降温至0℃,将体系保持搅拌并保持外置冰水浴,缓慢加入5.0g的60%的氢化钠(124.6mmol,2.75eq),加入完毕后滴加4.0g的2-氟磺酰基二氟乙酸(16.1mmol,2eq),用至少10min滴加完毕,撤掉冰水浴缓慢升至常温,持续反应直至用hplc与lcms监测反应完毕后,滴加20ml饱和nh4cl溶液与30mlh2o淬灭氢化钠,溶液逐渐变为澄清的黑褐色液体并析出固体,全部抽滤,再向混合液中加入600ml乙酸乙酯进行萃取并分离有机相,再用每次100ml饱和氯化钠溶液洗涤有机相2次,有机相干燥旋干,5倍拌样质量拌样,石油醚:乙酸乙酯=7:1的前提下200-300硅胶过柱,得2-二氟甲氧基-5-溴吡啶-3-甲酸甲酯9.2g,收率为72.1%,为白色粉末状固体。
检测数据为,1hnmr(400hz,dmso):δppm8.63(d,j=2.0hz,1h),8.47(d,j=2.0hz,1h),7.72(t,jh-f=71.6hz,1h),3.86(s,3h).lcms:(retentiontime:3.908min):283.0[m+1]。
(3)将10.0g的2-二氟甲氧基-5-溴吡啶-3-甲酸甲酯溶于50ml水与50ml四氢呋喃的混合液中,用外置冰水浴将体系降温至约10℃,向体系中缓慢加入4.5g的一水合氢氧化锂(106.5mmol,3eq),至少2min加入完毕,反应至少2.5h后,通过hplc与lcms监测反应完毕后,向体系中滴加12n的hcl直至将ph调至约2,然后每次加入100ml二氯甲烷对混合物进行萃取2次,合并有机相干燥旋干,得2-二氟甲氧基-5-溴吡啶-3-甲酸8.5g,收率为85.3%,为白色固体。
检测数据为,lcms:(retentiontime:3.223min):268.0[m+1]。
(4)将8.5g的2-二氟甲氧基-5-溴吡啶-3-甲酸全部加入到80ml三氯甲烷中,用外置冰水浴将体系降温至0-5℃,然后滴入16ml氯化亚砜,滴完后将体系升温至50℃反应30分钟,将反应混合物全部旋干,将残余物溶于50ml四氢呋喃,然后滴入100ml预冷至0-10℃之间的氨水,将体系自然升至室温,反应12h,旋掉全部溶剂,析出固体,抽滤,用50ml水淋洗,烘干,得到2-二氟甲氧基-5-溴吡啶-3-甲酰胺7.2g,收率84.7%。
(5)将7.2g的2-二氟甲氧基-5-溴吡啶-3-甲酰胺加入到100ml二氯甲烷中,然后加入21ml三乙胺,用外置冰水浴将反应混合物降温至0-5℃,滴入14g三氟乙酸酐,滴完后将反应混合物自然升至室温,反应16小时,倒入100ml冰水混合物中,分液,水相用50ml二氯甲烷萃取一次,然后有机相再用50ml饱和食盐水洗一次,无水硫酸钠干燥,抽滤,旋干得2-二氟甲氧基-3-氰基-5-溴吡啶3.4g,收率50.8%。
(6)在氮气保护下,将2.5g的2-二氟甲氧基-3-氰基-5-溴吡啶(10mmol)溶于30ml四氢呋喃种,用外置冰水浴将反应混合物降温至0℃,滴加入40ml硼烷四氢呋喃溶液,滴加完毕后自然升至室温,保温反应1h,再升温至出现回流,保持反应过夜,中途降温取样,将取出的样品慢慢滴加到甲醇和水中,送hplc-lcms监测反应完毕后,将反应混合物降至0℃,慢慢滴加入70ml甲醇,滴加完毕后,慢慢升至室温,搅拌至少1h,然后回流至少1h,除去溶剂得到2.39g油状液体2-二氟甲氧基-5-溴吡啶-3-甲胺。
(7)将2.39g的2-二氟甲氧基-5-溴吡啶-3-甲胺溶于30ml甲基叔丁基醚,将混合物用冰盐浴降温至-10℃,滴加入7n的二氧六环/hcl(2.5eq),搅拌反应至少1h,抽滤得到2-二氟甲氧基-5-溴吡啶-3-甲胺盐酸盐固体2.3g。
(8)将2.3g的2-二氟甲氧基-5-溴吡啶-3-甲胺盐酸盐在20ml二氯甲烷内充分分散,在低速搅拌下加入3.0g三乙胺,保持常温下分批加入3.3g的boc酸酐(15mmol),反应3h,加入10ml水,10mlx3二氯甲烷分液,盐水洗,干燥,除去溶剂,柱分离得到boc保护2-二氟甲氧基-5-溴吡啶-3-甲胺950mg,为黄色油状物,(6)-(8)三步收率为26.9%。
(9)在搅拌下,将4.0g的boc保护2-二氟甲氧基-5-溴吡啶-3-甲胺(11.3mmol)溶于120ml二氧六环中,在室温下将5.76g的双戊酰二硼(22.6mmol,2eq)与3.33g的乙酸钾(33.9mmol,3eq)缓慢加入至体系中,保持搅拌时体系充分分散,将体系抽冲氮气三次后,在氮气保护下向体系中加入464mg的pd(dppf)cl2-ch2cl2(0.568mmol,0.05eq),升温至85℃回流后保持温度,反应至少12h,hplc监测反应完毕后,冷却至室温,加入360ml乙酸乙酯稀释,抽滤后旋干拌样,石油醚:乙酸乙酯=20:1条件下,5倍拌样200-300硅胶过柱,得到3g产品,用30ml石油醚打浆30min,抽滤,得到纯品((2-二氟甲氧基-5-频那醇酯基)吡啶-3-基)甲基)氨基甲酸叔丁酯2.5g,收率为55.2%,为白色固体。
检测数据为,1hnmr(400hz,cdcl3):δppm8.43(d,j=1.6hz,1h),8.04(s,1h),7.57(t,jh-f=72.4hz,1h),4.97(brs,1h),4.32(d,j=5.2hz,2h),1.45(s,9h),1.33(s,12h)。lcms(retentiontime:3.277+4.589min):400.9([m+1]+)。
(10)将20.8g的nabo3-4h2o(135mmol,3eq)溶于40mlh2o中,用外置冰水浴降温至0℃,将18g((2-二氟甲氧基-5-频那醇酯基)吡啶-3-基)甲基)氨基甲酸叔丁酯(45mmol)溶于360ml四氢呋喃后滴加进体系,滴加完毕后升至室温,至少反应2h后,向体系中加入400ml饱和氯化铵与400ml乙酸乙酯,分液后,水相用400ml乙酸乙酯再次萃取,有机相用400mlh2o洗涤后,干燥,旋干拌样,石油醚:乙酸乙酯=10:1条件下,5倍拌样200-300硅胶过柱,得产物((2-(二氟甲氧基)-5-羟基吡啶-3-基)甲基)氨基甲酸叔丁酯10g,收率为76.6%,为浅黄色固体。
检测数据为,1hnmr(400hz,cdcl3):δppm7.67(s,1h),7.35(t,jh-f=73.6hz,1h),7.23(d,j=2.4hz,1h),5.19(brs,1h),4.24(d,j=6.0hz,2h),1.45(s,9h)。lcms(retentiontime:3.409min):290.9[m+0.7]。
经计算,总收率为2.26%,我们用同等条件,只是萃取使用普通器皿进行萃取,其他条件完全一样,结果收率只有1.83%,明显较低,而且时间要长一半左右。
实施例5
前述(1)-(10)中的萃取,利用所述的超声萃取装置实施,具体步骤为。
(a)准备步骤:清洗并晾干萃取部,将萃取部完全套入外壳部内固定,再将外壳部套入圆环部内,以使得周缘(24)得以被圆环部(42)上部承托,将振头槽(27)左侧顶入主壳(22)上的凹坑内,使用电工胶布在振头槽高摄氏度多圈缠绕,将其固定在主壳(22)上。该凹坑的深度可为5mm,该凹坑可以与振头槽左侧紧贴适配,插入即可初步固定,减轻胶布的固定压力,该振头槽左侧可以进一步具有与凹坑内的特定凸部配合的凹部,以增强固定效果。电工胶布可以由一条方便拆装的皮带替代,拆装更加容易,固定效果更好。
(b)盖上步骤:打开上盖(13),将欲萃取的混合物和溶剂倒入,放置破乳组件使球部在上开口之外且锥部向内,小心盖上上盖。上盖上可以另设放气孔,放气孔配有放气阀,放气阀在提拉到一定程度时可以允许气体通过,用于调节内外气压。破乳组件可以布置多个锥部高低不同的多个。
(c)震动晃动步骤:推动外壳部,使外壳部钟摆式前后晃动16次,手动空直外壳部大致静止,启动超声装置并使得振头以30khz的频率震动2min,静置2min。
(d)重复步骤:重复步骤c4次。每次重复开始前可以经观察槽确认基本稳定分层。
(e)接取步骤:停止萃取,打开上盖,拿出破乳组件,用不同的玻璃器分别在出液口下方接取不同相态的混合物,萃取部的最后一种相态可从上口倒出。
实施例6
前述(1)-(10)中的萃取,具体步骤为。
(a)准备步骤:清洗并晾干萃取部,将萃取部完全套入外壳部内固定,再将外壳部套入圆环部内,以使得周缘(24)得以被圆环部(42)上部承托,将振头槽(27)左侧顶入主壳(22)上的凹坑内,使用电工胶布在振头槽高摄氏度多圈缠绕,将其固定在主壳(22)上。该凹坑的深度可为7mm,该凹坑可以与振头槽左侧紧贴适配,插入即可初步固定,减轻胶布的固定压力,该振头槽左侧可以进一步具有与凹坑内的特定凸部配合的凹部,以增强固定效果。电工胶布可以由一条方便拆装的皮带替代,拆装更加容易,固定效果更好。
(b)盖上步骤:打开上盖(13),将欲萃取的混合物和溶剂倒入,放置破乳组件使球部在上开口之外且锥部向内,小心盖上上盖。上盖上可以另设放气孔,放气孔配有放气阀,放气阀在提拉到一定程度时可以允许气体通过,用于调节内外气压。破乳组件可以布置多个锥部高低不同的多个。
(c)震动晃动步骤:推动外壳部,使外壳部钟摆式前后晃动20次,手动空直外壳部大致静止,启动超声装置并使得振头以45khz的频率震动2min,静置4min。
(d)重复步骤:重复步骤c5次。每次重复开始前可以经观察槽确认基本稳定分层。
(e)接取步骤:停止萃取,打开上盖,拿出破乳组件,用不同的玻璃器分别在出液口下方接取不同相态的混合物,萃取部的最后一种相态可从上口倒出。
优选地,前述所有试剂均为化学纯以上纯度,或者均为优级纯。所述水均为去离子水,优选地为双蒸水。上述剂量配比选择均是最初实验室制得的数据,实际制备中,为了量大好制,一般是至少乘10倍剂量起算的,则此时普通萃取设备不敷使用,需要专门萃取设备,应对大剂量萃取。
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何不经过创造性劳动想到的变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应该以权利要求书所限定的保护范围为准。
- 还没有人留言评论。精彩留言会获得点赞!