一种杂蒽衍生物及其应用和有机电致发光器件的制作方法
[0001]
本发明属于光电材料领域,具体涉及一种杂蒽衍生物及其应用和有机电致发光器。
背景技术:
[0002]
oled,即有机发光二极管,又称为有机电激光显示。oled具有自发光的特性,采 用非常薄的有机材料涂层和玻璃基板,当电流通过时,有机材料就会发光,而且oled 显示屏幕可视角度大,并且能够显著节省电能,因此oled被视为21世纪最具前途的产 品之一。但迄今为止,oled器件仍未能实现普及化应用,其中,器件的效率是制约其 普及化的重要原因。
[0003]
在光学器件中,可在器件的表面蒸镀介质薄膜来降低表面的反射损耗,原理是薄膜 的干涉相消作用,进一步可以解释为光波在传播的过程中,由于边界条件的不同,在两 种不同传播介质的界面处,能量的分布发生了变化。因此,在有机电致发光器件的出光 侧金属电极之外引入出光层材料,它对器件的电学性能基本不会造成影响,只是改变光 波的透射和反射能量的分布,增强光的输出耦合能力。作为微腔oled的出光耦合层材 料,主要考虑到它们的高折射率指数、可见光范围内对光的低吸收率以及比较容易的蒸 发生长方式等特性,增强光的耦合输出能力,提高器件外量子效率,减少光在器件内部 的损耗。
[0004]
目前,光电领域虽然开发了一些alq3、tpbi及其它高折射低吸收的出光层材料,但 至今,还没有一种材料能满足产业层面对出光层的要求,在器件内对光的耦合输出能力 有限。开发出不影响器件本征性能的同时,大幅度提高器件出光效率的出光层材料依旧 是光电领域研发人员追求的目标之一。
技术实现要素:
[0005]
本发明要解决的技术问题是:提供一种对器件出光效率提升幅度更高的出光层材料, 从而将其应用至有机电致发光器件,以更好的解决有机材料和基底间的折射率差异致使 有机电致发光器件内全反射造成的光的损耗。
[0006]
本发明提供了一种杂蒽衍生物,杂蒽衍生物为式(i)所示结构:
[0007][0008]
其中,x独立的选自o、s、s=o或s(=o)2中的一种;
[0009]
r1、r2分别独立的选自下列a1、a2、a3、a4、a5或a6所示的基团中的一种:
[0010][0011]
其中,z
1-z5分别独立的选自cr6或n;
[0012]
q
1-q8分别独立的选自cr7或n;
[0013]
l
1-l6分别独立的选自单键,未取代的或由氟基、硝基、氰基、c
1-20
的烷基取代的亚 苯基,未取代的或由氟基、硝基、氰基、c
1-20
的烷基取代的亚联苯基,未取代的或由氟 基、硝基、氰基、c
1-20
的烷基取代的亚萘基中的一种;
[0014]
ar1、ar2、ar3分别独立的单环芳烃、多环芳烃或稠环芳烃,所述ar1、ar2、ar3与连 接的萘环之间直接稠合,各所述虚线分别独立的表示ar1、ar2和ar3存在或不存在;
[0015]
y1、y2分别独立的选自c(r8)(r9)、n(r
10
)、o、s,r9、r
10
相互独立或者通过单 键连接;
[0016]
r
3-r
10
分别独立的选自氢、氟基、硝基、氰基、磺酰基、c
1-20
的烷基、c
1-20
的硅烷基、 c
6-50
的芳基、c
3-50
的杂芳基、c
6-50
的芳氧基或c
6-50
的芳硫基中的一种;
[0017]
*为取代位点。
[0018]
在上述方案的基础上,本发明该可以进行如下改进:
[0019]
进一步,所述c
1-20
的烷基选自甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲 丁基或叔丁基中的一种;c
6-50
的芳基选自:取代基取代或未取代的苯基、取代基取代或 未取代的联苯基、由取代基取代或未取代的三联苯基、取代基取代或未取代的萘基、取 代基取代或未取代的蒽基、取代基取代或未取代的菲基、取代基取代或未取代的芘基、 取代基取代或未取代的苯并菲基、取代基取代或未取代的芴基、取代基取代或未取代的 螺二芴基中的一种;c
3-50
的杂芳基选自:取代基取代或未取代的吡啶基、取代基取代或 未取代的嘧啶基、取代基取代或未取代的吡嗪基、取代基取代或未取代的三嗪基、取代 基取代或未取代的吲哚基、取代基取代或未取代的苯并呋喃基、取代基取代或未取代的 苯并噻吩基、取代基取代或未取代的苯并噁唑基、取代基取代或未取代的苯并噻唑基、 取代基取代或未取代的咔唑基、取代基取代或未取代的二苯并呋喃基、取代或未取代的 二苯并噻吩基中的一种。
[0020]
进一步,取代基选自:氰基、氟基、硝基、甲基、乙基、正丙基、异丙基、正丁基、 异丁基、仲丁基、叔丁基、苯基、联苯基、三联苯基、萘基、蒽基、芴基、吡啶基、嘧 啶基、吡嗪基、三嗪基、吲哚基、苯并呋喃基、苯并噻吩基、苯并噁唑基、苯并噻唑基、 咔唑基、二苯并呋喃基或二苯并噻吩基中的一种。
[0021]
优选的,a1选自下列基团:
[0022][0023]
a2选自下列基团:
[0024][0025]
a3选自下列基团:
[0026]
[0027][0028]
a4选自下列基团:
[0029][0030]
a5选自下列基团:
[0031]
[0032]
a6选自下列基团:
[0033][0034]
进一步的,所述r1和r2分别连接在杂蒽1位和6位,或者1位和9位,或者2位和 8位,或者2位和7位;
[0035]
优选的,所述r1和r2分别连接在杂蒽的2位和7位。
[0036]
优选的,所述x分别独自的选自s、s=o、s(=o)2中的一种。
[0037]
基于本发明的杂蒽衍生物,在杂蒽的2,7位采用不对称修饰,可强化非金属重原子 s在杂蒽衍生物的重原子效应,使得杂蒽衍生物具有更高的玻璃化转变温度和高折射低 吸收特性。
[0038]
优选的,杂蒽衍生物选自以下结构:
[0039]
[0040]
[0041]
[0042]
[0043]
[0044]
[0045]
[0046]
[0047]
[0048]
[0049]
[0050]
[0051]
[0052]
[0053][0054]
在杂蒽基团的2,7位设计特定基团,从而构建得到的本申请中的杂蒽衍生物表现出 了优异的出光层材料特性,将其作为出光层材料制备得到的有机电致发光器件,相比于 同样结构的没有出光层的有机电致发光器件以及同样结构利用传统的出光层材料,比如 alq3,在电流效率、外量子效率上具有显著的优势;上述杂蒽衍生物在杂蒽的2,7位修 饰特定基团后,由于偏向于线型的空间构型,使得材料蒸镀成膜后定向排列,微观状态 下的膜表面更为光滑,有利于作为出光层增强oled器件光的耦合输出能力,使得本发 明的杂蒽衍生物作为出光层制备的器件,在电流效率、外量子效率(出光效率)上具有 显著的优势,是一种优异的出光层材料。
[0055]
本发明第二个方面提供了一种杂蒽衍生物的应用,用于有机电致发器件的出光层
材 料。
[0056]
本发明第三个方面提供了一种有机电致发光器件,包括:出光层、阴极、阳极以及 阴极和阳极之间的有机层,其特征在于,所述出光层形成在所述阴极远离所述阳极的一 侧表面,所述出光层由如上所述杂蒽衍生物形成。
[0057]
基于本发明的杂蒽衍生物,具有优异的出光特性,可作为有机电致发光器件的出光 层材料,对器件出光效率具有显著提升;且该出光层材料具有优异的成膜特性,能更好 的解决有机材料和基底间的折射率差异致使有机电致发光器件内全反射造成的光的损 耗。
[0058]
进一步的,该有机电致发光器件出光层的厚度为1-120nm。
附图说明
[0059]
本发明的上述和/或附加的方面和优点从结合下面附图对实施例的描述中将变得明 显和容易理解,其中:
[0060]
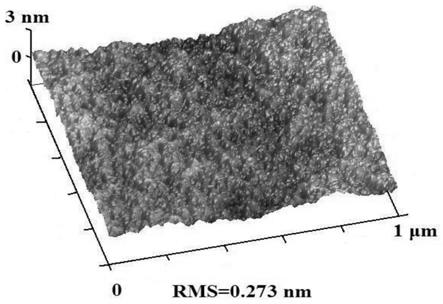
图1为基于本发明化合物94蒸镀得到的出光层膜层的原子力显微镜成像图;
[0061]
图2为对比物2蒸镀得到的出光层膜层的原子力显微镜成像图。
具体实施方式
[0062]
下面详细描述本发明的实施例。下面描述的实施例是示例性的,仅用于解释本发明, 而不能理解为对本发明的限制。实施例中未注明具体技术或条件的,按照本领域内的文 献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者, 均为可以通过市购获得的常规产品。
[0063]
实施例1
[0064]
化合物21的制备
[0065][0066]
s1.在250ml三口瓶中,投入咔唑(5.0g,30mmol)、1,4-二溴萘(11.44g,40mmol)、 叔丁醇钠(5.76g,60mmol)、三叔丁基膦四氟硼酸盐(0.087g,0.3mmol),按照咔唑的物质的 量计,加入3-5倍体积的甲苯,在氮气氛围下,加入三(二亚苄基丙酮)二钯 (0.15g,0.15mmol),100-115℃下反应4-16h,液相监测反应基本完成,冷却至室温,水洗, 分液,有机相浓缩后与滤饼一起,用乙醇、乙酸乙酯或其组合物打浆或重结晶纯化1-3 次,即可得到7.59g 9-(4-溴萘-1-基)-9h-咔唑,收率68%;
[0067]
s2.在250ml三口瓶中,投入9-(4-溴萘-1-基)-9h-咔唑(5.58g,15mmol),加入40-60ml 干燥的thf,冷却至-78℃时,缓慢加入7.5ml正丁基锂(2.4m)(18mmol),反应1 小时
后,接着加入硼酸三异丙酯(4.23g,22.5mmol),继续反应2小时,升至室温后反应 一晚,冷却至0℃后,加入hcl(2.0m)40ml水解反应后,用二氯甲烷萃取,蒸馏除去溶 剂后,得到4.0g(4-(9h-咔唑-9-基)萘-1-基)硼酸,产率79%。
[0068]
s3.在250ml的三口瓶中,投入2,4-二苯基-6-硼酸频哪醇酯吡啶(7.14g,20mmol)、 2,7-二溴噻蒽(9.35g,25mmol)、碳酸钾(5.53g,40mmol),按照2,4-二苯基-6-频哪醇酯吡 啶的物质的量计,加入2-4倍体积的甲苯、1-2倍体积的乙醇和1-2倍体积的水,在氮气 氛围下,加入四(三苯基膦)钯(0.05g,0.04mmol),升温至70-90℃反应6-18h,液相监测反 应完成,冷却至室温,反应液过滤,有机相浓缩后与滤饼一起用乙醇、乙酸乙酯或其组 合物打浆或重结晶纯化1-3次,即可得到6.81g 2-(7-溴噻蒽-2-基)-4,6-二苯基吡啶,收率 65%;
[0069]
s4.在100ml的三口瓶中,投入(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.39g,10mmol)、2-(7
-ꢀ
溴噻蒽-2-基)-4,6-二苯基吡啶(5.24g,10mmol)、碳酸钾(2.76g,20mmol),按照(4-(二苯基 胺基)萘-1-基)硼酸的物质的量计,加入2-3倍体积的甲苯、1-1.5倍体积的乙醇和1-1.5倍 体积的水,在氮气氛围下,加入四(三苯基膦)钯(0.03g,0.02mmol),升温至70-90℃反应 6-18h,液相监测反应完成,冷却至室温,反应液过滤,有机相浓缩后与滤饼一起拌样过 硅胶柱,用10:1的石油醚与二氯甲烷的混合液淋洗,淋洗液浓缩,即可得到5.47g目标 化合物(21),收率74%。
[0070]
质谱仪maldi-tof-ms(m/z)=736.9518,理论分子量:736.9510;元素分析:理论 值c
51
h
32
n2(%):c 83.12,h 4.38,n 3.80;实测值:c 82.13,h 4.35,n 3.80。
[0071]
实施例2
[0072]
化合物29的制备
[0073]
s1.将合成实施例1中s3的2,4-二苯基-6-硼酸频哪醇酯吡啶(7.14g,20mmol)替换 为2-([1,1'-联苯]-3-基)硼酸频哪醇酯(5.60g,20mmol),其他合成步骤同合成实施例1 中的s3,即可得到6.08g 2-([1,1'-联苯]-3-基)-7-溴噻蒽,收率68%;
[0074]
s2.将合成实施例1中s4的(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.39g,10mmol)替换为 2-(二苯并呋喃-4-基)-4-苯基-6-(硼酸频那醇酯-2-基)-1,3,5-三嗪(4.49g,10mmol),2-(7-溴 噻蒽-2-基)-4,6-二苯基吡啶(5.24g,10mmol)替换为2-([1,1'-联苯]-3-基)-7-溴噻蒽(4.47 g,10mmol),其他合成步骤同合成实施例1中的s4,即可得到4.82g目标化合物(29), 收率70%。
[0075]
质谱仪maldi-tof-ms(m/z)=689.8519,理论分子量:689.8510;元素分析:理论 值c
45
h
27
n3(%):c 78.35,h 3.95,n 6.09;实测值:c 78.33,h 3.97,n 6.10。
[0076]
实施例3
[0077]
化合物33的制备
[0078]
s1.将合成实施例1中s3的2,4-二苯基-6-硼酸频哪醇酯吡啶(7.14g,20mmol)替换 为2-([1,1':3',1
”-
三联苯]-5'-基)硼酸频哪醇酯(7.12g,20mmol),其他合成步骤同合成实 施例1中的s3,即可得到7.00g 2-([1,1':3',1
”-
三联苯]-5'-基)-7-溴噻蒽,收率67%;
[0079]
s2.将合成实施例1中s4的(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.39g,10mmol)替换为 9-苯基-3-(硼酸频哪醇酯-2-基)-9h-咔唑(3.69g,10mmol),2-(7-溴噻蒽-2-基)-4,6-二苯基 吡啶(5.24g,10mmol)替换为2-([1,1':3',1
”-
三联苯]-5'-基)-7-溴噻蒽(5.24g,10mmol), 其他合成步骤同合成实施例1中的s4,即可得到5.21g目标化合物(33),收率
76%。
[0080]
质谱仪maldi-tof-ms(m/z)=685.9026,理论分子量:685.9030;元素分析:理论 值c
48
h
31
n(%):c 84.05,h 4.56,n 2.04;实测值:c 84.04,h 4.59,n 2.03。
[0081]
实施例4
[0082]
化合物49的制备
[0083]
s1.将合成实施例1中s3的2,4-二苯基-6-硼酸频哪醇酯吡啶(7.14g,20mmol)替换 为2-(萘-1-基)-4苯基-6-(硼酸频哪醇酯-2-基)-1,3,5-三嗪(8.19g,20mmol),其他合成步骤 同合成实施例1中的s3,即可得到7.26g 2-(7-溴噻蒽-2-基)-4-(萘-1-基)-6-苯基-1,3,5-三嗪, 收率63%;
[0084]
s2.将合成实施例1中s4的(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.39g,10mmol)替换为(8
-ꢀ
苯基二苯并呋喃-2-基)硼酸频哪醇酯(3.70g,10mmol),2-(7-溴噻蒽-2-基)-4,6-二苯基吡啶 (5.24g,10mmol)替换为2-(7-溴噻蒽-2-基)-4-(萘-1-基)-6-苯基-1,3,5-三嗪(5.76 g,10mmol),其他合成步骤同合成实施例1中的s4,即可得到5.18g目标化合物(49), 收率70%。
[0085]
质谱仪maldi-tof-ms(m/z)=739.9105,理论分子量:739.9110;元素分析:理论 值c
49
h
29
n3(%):c 79.54,h 3.95,n 5.68;实测值:c 79.54,h 3.98,n 5.66。
[0086]
实施例5
[0087]
化合物55的制备
[0088]
s1.将合成实施例1中s3的2,4-二苯基-6-硼酸频哪醇酯吡啶(7.14g,20mmol)替换 为苯硼酸(2.44g,20mmol),其他合成步骤同合成实施例1中的s3,即可得到4.68g 2
-ꢀ
溴-7-苯基噻蒽,收率63%;
[0089]
s2.将合成实施例1中s4的(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.39g,10mmol)替换为 (9,9-二甲基-7-苯基l-9h-芴-2-基)硼酸(3.14g,10mmol),2-(7-溴噻蒽-2-基)-4,6-二苯基吡 啶(5.24g,10mmol)替换为2-溴-7-苯基噻蒽(3.71g,10mmol),其他合成步骤同合成实 施例1中的s4,即可得到4.03g目标化合物(55),收率72%。
[0090]
质谱仪maldi-tof-ms(m/z)=560.7718,理论分子量:560.7730;元素分析:理论 值c
39
h
28
(%):c 83.53,h 5.03;实测值:c 83.52,h 5.05。
[0091]
实施例6
[0092]
化合物64的制备
[0093]
s1.将合成实施例1中s3的2,4-二苯基-6-硼酸频哪醇酯吡啶(7.14g,20mmol)替换 为4-叔丁基苯硼酸(3.56g,20mmol),其他合成步骤同合成实施例1中的s3,即可得到 5.55g2-溴-7-(4-叔丁基苯基)噻蒽,收率65%;
[0094]
s2.在100ml三口瓶中,投入3,6-二苯基-9h-咔唑(3.19g,10mmol)、2-溴-7-(4-叔 丁基苯基)噻蒽(4.27g,10mmol)、叔丁醇钠(1.92g,20mmol)、三叔丁基膦四氟硼酸盐 (0.029g,0.1mmol),按照3,6-二苯基-9h-咔唑的物质的量计,加入3-6倍体积的甲苯,在 氮气氛围下,加入三(二亚苄基丙酮)二钯(0.048g,0.05mmol),100-115℃下反应4-16h,液相 监测反应基本完成,冷却至室温,反应液过滤,有机相浓缩后与滤饼一起拌样过硅胶柱, 用10:1的石油醚与二氯甲烷的混合液淋洗,淋洗液浓缩,即可得到5.26g目标化合物 (64),收率79%;
[0095]
质谱仪maldi-tof-ms(m/z)=665.9128,理论分子量:665.9130;元素分析:理论 值c
46
h
35
n(%):c 82.97,h 5.30,n 2.10;实测值:c 82.95,h 5.31,n 2.12。
[0096]
实施例7
[0097]
化合物68的制备
[0098]
s1.将合成实施例6中s1的4-叔丁基苯硼酸(3.56g,20mmol)替换为4-([1,1'-联苯]-3
-ꢀ
基)-2-苯基-6-硼酸频哪醇酯嘧啶(8.69g,20mmol),其他合成步骤同合成实施例6中的 s1,即可得到7.46g4-([1,1'联苯]-3-基)-6-(7-溴噻蒽-2-基)-2-苯基嘧啶,收率62%;
[0099]
s2.将合成实施例6中s2中的3,6-二苯基-9h-咔唑(3.19g,10mmol)替换为咔唑 (1.67g,10mmol),2-溴-7-(4-叔丁基苯基)噻蒽(4.27g,10mmol)替换为4-([1,1'联苯]-3
-ꢀ
基)-6-(7-溴噻蒽-2-基)-2-苯基嘧啶(6.01g,10mmol),其他合成步骤同合成实施例6中的 s2,即可得到5.57g目标化合物(68),收率81%
[0100]
质谱仪maldi-tof-ms(m/z)=687.8802,理论分子量:687.8790;元素分析:理论 值c
46
h
29
n3(%):c 80.32,h 4.25,n 6.11;实测值:c 80.30,h 4.28,n 6.11。
[0101]
实施例8
[0102]
化合物74的制备
[0103]
s1.将合成实施例1中s3的2,4-二苯基-6-硼酸频哪醇酯吡啶(7.14g,20mmol)替换 为苯硼酸(2.44g,20mmol),其他合成步骤同合成实施例1中的s3,即可得到4.68g 2
-ꢀ
溴-7-苯基噻蒽,收率63%;
[0104]
s2.将合成实施例1中s4的(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.39g,10mmol)替换为 (4-(9h-[3,9'-二咔唑]-9-基)苯基)硼酸(4.52g,10mmol),2-(7-溴噻蒽-2-基)-4,6-二苯基吡啶 (5.24g,10mmol)替换为2-溴-7-苯基噻蒽(3.71g,10mmol),其他合成步骤同合成实施 例1中的s4,即可得到5.10g目标化合物(74),收率73%。
[0105]
质谱仪maldi-tof-ms(m/z)=698.9013,理论分子量:698.9020;元素分析:理论 值c
48
h
30
n2(%):c 82.49,h 4.33,n 4.01;实测值:c 82.50,h 4.35,n 4.00。
[0106]
实施例9
[0107]
化合物88的制备
[0108]
s1.将合成实施例1中s3的2,4-二苯基-6-硼酸频哪醇酯吡啶(7.14g,20mmol)替换 为2,4-二苯基-6-硼酸频哪醇酯-1,3,5三嗪(7.18g,20mmol),其他合成步骤同合成实施 例1中的s3,即可得到7.05g2-(7溴噻蒽-2-基)-4,6-二苯基-1,3,5-三嗪,收率67%;
[0109]
s2.将合成实施例1中s4的(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.39g,10mmol)替换为 3-(1-苯基-1h-苯并咪唑-2-基)苯硼酸频哪醇酯(3.96g,10mmol),2-(7-溴噻蒽-2-基)-4,6
-ꢀ
二苯基吡啶(5.24g,10mmol)替换为2-(7溴噻蒽-2-基)-4,6-二苯基-1,3,5-三嗪(5.26 g,10mmol),其他合成步骤同合成实施例1中的s4,即可得到5.15g目标化合物(88), 收率72%。
[0110]
质谱仪maldi-tof-ms(m/z)=715.8935,理论分子量:715.8930;元素分析:理论 值c
46
h
29
n5(%):c 77.18,h 4.08,n 9.78;实测值:c 77.17,h 4.10,n 9.78。
[0111]
实施例10
[0112]
化合物94的制备
值c
50
h
31
n(%):c 84.59,h 4.40,n 1.97;实测值:c 84.62,h 4.40,n 1.95。
[0131]
实施例14
[0132]
化合物122的制备
[0133]
s2.将合成实施例1中s3的2,4-二苯基-6-硼酸频哪醇酯吡啶(7.14g,20mmol)替换 为(10-苯基蒽-9-基)硼酸(5.96g,20mmol),其他步骤同合成实施例1中s3,即可得到 6.90g 2-溴-7-(10-苯基蒽-9-基)噻蒽,收率63%;
[0134]
s3.将合成实施例1中s4的(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.39g,10mmol)替换为 二苯并噻吩-2-基硼酸(2.28g,10mmol),2-(7-溴噻蒽-2-基)-4,6-二苯基吡啶(5.24g,10mmol) 替换为2-溴-7-(10-苯基蒽-9-基)噻蒽(5.47g,10mmol),其他合成步骤同合成实施例1中 的s4,即可得到4.88g目标化合物(122),收率75%。
[0135]
质谱仪maldi-tof-ms(m/z)=650.8708,理论分子量:650.8720;元素分析:理论 值c
44
h
26
(%):c 81.20,h 4.03;实测值:c 81.18,h 4.03。
[0136]
实施例15
[0137]
化合物126的制备
[0138]
s1.将合成实施例1中s3的2,4-二苯基-6-硼酸频哪醇酯吡啶(7.14g,20mmol)替换 为(3-(萘-2-基)苯基)硼酸(4.96g,20mmol),其他步骤同合成实施例1中s3,即可得到 6.36g 2-溴-7-(3-(萘-2-基)苯基)噻蒽,收率64%;
[0139]
s2.将合成实施例1中s4的(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.39g,10mmol)替换为 (3-(9h-咔唑-9-基)苯基)硼酸(2.87g,10mmol),2-(7-溴噻蒽-2-基)-4,6-二苯基吡啶(5.24 g,10mmol)替换为2-溴-7-(3-(萘-2-基)苯基)噻蒽(4.97g,10mmol),其他合成步骤同合成 实施例1中的s4,即可得到5.08g目标化合物(126),收率77%。
[0140]
质谱仪maldi-tof-ms(m/z)=659.8644,理论分子量:659.8650;元素分析:理论 值c
46
h
29
n(%):c 83.73,h 4.43,n 2.12;实测值:c 83.70,h 4.45,n 2.12。
[0141]
实施例16
[0142]
化合物150的制备
[0143]
s1.将合成实施例1中s3的2,4-二苯基-6-硼酸频哪醇酯吡啶(7.14g,20mmol)替换 为(3-(萘-1-基)苯基)硼酸(4.96g,20mmol),其他步骤同合成实施例1中s3,即可得到 6.17g 2-溴-7-(3-(萘-2-基)苯基)噻蒽,收率62%;
[0144]
s2.将合成实施例1中s4的(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.39g,10mmol)替换为 (3-(1-苯基-1h-苯并咪唑-2-基)苯基)硼酸(3.14g,10mmol),2-(7-溴噻蒽-2-基)-4,6-二苯基 吡啶(5.24g,10mmol)替换为2-溴-7-(3-(萘-1-基)苯基)噻蒽(4.96g,10mmol),其他合成 步骤同合成实施例1中的s4,即可得到5.35g目标化合物(150),收率78%。
[0145]
质谱仪maldi-tof-ms(m/z)=686.8903,理论分子量:686.8910;元素分析:理论 值c
47
h
30
n2(%):c 82.18,h 4.40,n 4.08;实测值:c 82.18,h 4.42,n 4.05。
[0146]
实施例17
[0147]
化合物154的制备
[0148]
将合成实施例10中步骤s2的(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.37g,10mmol)替换为(9-苯基-9h-咔唑-3-基)硼酸(2.87g,10mmol),其他合成过程同合成实施例步骤s2,即可 得到2.38g目标化合物(154),收率68%。
[0149]
质谱仪maldi-tof-ms(m/z)=698.9027,理论分子量:698.9020;元素分析:理论 值c
48
h
30
n2(%):c 82.49,h 4.33,n 4.01;实测值:c 82.50,h 4.31,n 4.00。
[0150]
实施例18
[0151]
化合物159的制备
[0152]
s1.将合成实施例6中s1的4-叔丁基苯硼酸(3.56g,20mmol)替换为(9-苯基-9h-咔 唑-3-基)硼酸(5.74g,20mmol),其他合成步骤同合成实施例6中的s1,即可得到6.76g 3-(7-溴噻蒽-2-基)-9-苯基-9h-咔唑,收率63%;
[0153]
s2.将合成实施例6中s2中的3,6-二苯基-9h-咔唑(3.19g,10mmol)替换为3-苯基
ꢀ-
9h-咔唑(2.43g,10mmol),2-溴-7-(4-叔丁基苯基)噻蒽(4.27g,10mmol)替换为3-(7
-ꢀ
溴噻蒽-2-基)-9-苯基-9h-咔唑(5.36g,10mmol),其他合成步骤同合成实施例6中的s2, 即可得到5.52g目标化合物(159),收率79%
[0154]
质谱仪maldi-tof-ms(m/z)=698.9020,理论分子量:698.9010;元素分析:理论 值c
48
h
30
n2(%):c 82.49,h 4.33,n 4.01;实测值:c 82.48,h 4.32,n 4.02。
[0155]
实施例19
[0156]
化合物176的制备
[0157]
s1.将合成实施例1中s3的2,4-二苯基-6-硼酸频哪醇酯吡啶(7.14g,20mmol)替换 为二苯并噻吩-2-基硼酸(4.56g,20mmol),其他步骤同合成实施例1中s3,即可得到 6.20g 2-溴-7-(二苯并噻吩-2-基)噻蒽,收率65%;
[0158]
s2.将合成实施例1中s4的(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.39g,10mmol)替换为 (3-(9h-咔唑-9-基)苯基)硼酸(2.87g,10mmol),2-(7-溴噻蒽-2-基)-4,6-二苯基吡啶(5.24 g,10mmol)替换为2-溴-7-(二苯并噻吩-2-基)噻蒽(4.77g,10mmol),其他合成步骤同合 成实施例1中的s4,即可得到4.86g目标化合物(176),收率76%。
[0159]
质谱仪maldi-tof-ms(m/z)=639.8502,理论分子量:639.8490;元素分析:理论 值c
42
h
25
n(%):c 78.84,h 3.94,n 2.19;实测值:c 78.84,h 3.95,n 2.20。
[0160]
实施例20
[0161]
化合物185的制备
[0162]
s1.将合成实施例1中s3的2,4-二苯基-6-硼酸频哪醇酯吡啶(7.14g,20mmol)替换 为(9-苯基-9h-咔唑-3-基)硼酸(4.56g,20mmol),其他步骤同合成实施例1中s3,即可 得到6.76g 3-(7-溴噻蒽-2-基)-9-苯基-9h-咔唑,收率63%;
[0163]
s2.将合成实施例1中s4的(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.39g,10mmol)替换为 (3-(1-苯基-1h-苯并咪唑-2-基)苯基)硼酸(3.14g,10mmol),2-(7-溴噻蒽-2-基)-4,6-二苯基 吡啶(5.24g,10mmol)替换为3-(7-溴噻蒽-2-基)-9-苯基-9h-咔唑(5.36g,10mmol),其他 合成步骤同合成实施例1中的s4,即可得到5.30g目标化合物(185),收率73%。
[0164]
质谱仪maldi-tof-ms(m/z)=725.9275,理论分子量:725.9280;元素分析:理论 值c
49
h
31
n3(%):c 81.07,h 4.30,n 5.79;实测值:c 81.05,h 4.32,n 5.79。
[0165]
实施例21
[0166]
化合物191的制备
[0167][0168]
在100ml的三口瓶中,投入2,7-二溴噻蒽(3.74g,10mmol)、3-苯基-9h-咔唑 (5.35g,22mmol)、碳酸钾(4.15g,30mmol)、邻菲罗啉(0.2g,1mmol),按照2,7-二溴噻 吩的物质的量计,加入3-5倍体积的二甲基乙酰胺,在氮气氛围下,加入碘化亚铜 (0.2g,1mmol),升温至165℃反应12-24h,液相监测反应完成,冷却至室温,水洗,分液, 有机相浓缩,用10:1的石油醚与二氯甲烷的混合液淋洗,淋洗液浓缩,即可得到3.84g 目标化合物(191),收率55%。
[0169]
质谱仪maldi-tof-ms(m/z)=698.9011,理论分子量:698.9020;元素分析:理论 值c
48
h
30
n2(%):c 82.49,h 4.33,n 4.01;实测值:c 82.47,h 4.35,n 4.00。
[0170]
实施例22
[0171]
化合物201的制备
[0172][0173]
在100ml的三口瓶中,投入2,7-二溴噻蒽(3.74g,10mmol)、(3-(9h-咔唑-9-基)苯基) 硼酸(6.32g,22mmol)、碳酸钾(2.76g,20mmol),按照2,7-二溴噻吩的物质的量计,加入 2-3倍体积的甲苯、1-1.5倍体积的乙醇和1-1.5倍体积的水,在氮气氛围下,加入四(三 苯基膦)钯(0.03g,0.02mmol),升温至70-90℃反应6-18h,液相监测反应完成,冷却至室温, 反应液过滤,有机相浓缩后与滤饼一起拌样过硅胶柱,用10:1的石油醚与二氯甲烷的混 合液淋洗,淋洗液浓缩,即可得到4.26g目标化合物(201),收率61%。
[0174]
质谱仪maldi-tof-ms(m/z)=698.9028,理论分子量:698.9020;元素分析:理论 值c
48
h
30
n2(%):c 82.49,h 4.33,n 4.01;实测值:c 82.48,h 4.34,n 4.02。
[0175]
实施例23
[0176]
化合物212的制备
[0177]
s1.将合成实施例1中s3的2,4-二苯基-6-硼酸频哪醇酯吡啶(7.14g,20mmol)替换 为(3-(9h-咔唑-9-基)苯基)硼酸(5.74g,20mmol),其他步骤同合成实施例1中s3,即可 得到7.30g9-(3-(7-溴噻蒽-2-基)苯基)-9h-咔唑,收率68%;
[0178]
s2.将合成实施例1中s4的(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.39g,10mmol)替换为 (3-(1-苯基-1h-苯并咪唑-2-基)苯基)硼酸(3.14g,10mmol),2-(7-溴噻蒽-2-基)-4,6-二苯基 吡啶(5.24g,10mmol)替换为9-(3-(7-溴噻蒽-2-基)苯基)-9h-咔唑(5.36g,10mmol),其 他合成步骤同合成实施例1中的s4,即可得到5.15g目标化合物(212),收率71%。
[0179]
质谱仪maldi-tof-ms(m/z)=725.9294,理论分子量:725.9280;元素分析:理论 值c
49
h
31
n3(%):c 81.07,h 4.30,n 5.79;实测值:c 81.08,h 4.30,n 5.80。
[0180]
实施例24
[0181]
化合物217的制备
[0182]
将合成实施例22中的(3-(9h-咔唑-9-基)苯基)硼酸(6.32g,22mmol)替换为(2-苯基苯 并噻唑-6-基)硼酸(5.61g,22mmol),其他步骤同合成实施例22,即可得到3.56g目标化 合物(217),收率56%。
[0183]
质谱仪maldi-tof-ms(m/z)=634.8485,理论分子量:634.8480;元素分析:理论 值c
38
h
22
n2(%):c 71.89,h 3.49,n 4.41;实测值:c 71.90,h 3.46,n 4.42。
[0184]
实施例25
[0185]
化合物224的制备
[0186]
s1.将合成实施例1中s3的2,4-二苯基-6-硼酸频哪醇酯吡啶(7.14g,20mmol)替换 为(4-叔丁基苯基)硼酸(3.56g,20mmol),其他合成步骤同合成实施例1中的s3,即可 得到5.55g 2-溴-7-(4-叔丁基苯基)噻蒽,收率65%;
[0187]
s2.将合成实施例1中s4的(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.39g,10mmol)替换为 (10-(9h-咔唑-9-基)蒽-9-基)硼酸(3.87g,10mmol),2-(7-溴噻蒽-2-基)-4,6-二苯基吡啶 (5.24g,10mmol)替换为2-溴-7-(4-叔丁基苯基)噻蒽(4.27g,10mmol),其他合成步骤同 合成实施例1中的s4,即可得到4.76g9-(10-(7-(4-叔丁基苯基噻蒽-2-基)蒽-9-基)-9h-咔唑, 收率69%。
[0188]
s3.在100ml三口瓶中,加入s2的9-(10-(7-(4-叔丁基苯基噻蒽-2-基)蒽-9-基)-9h-咔 唑(3.45g,5mmol)、二氯甲烷(30ml)、双氧水(5ml)、醋酸(15ml),升温至70℃, 反应2-6h,液相监测原料基本无剩余,降温停止反应。用硅胶漏斗过滤反应液,过滤液 水洗,分层,浓缩,即可得到3.58g目标化合物(224),收率95%;
[0189]
质谱仪maldi-tof-ms(m/z)=753.9301,理论分子量:753.9310;元素分析:理论 值c
48
h
35
n(%):c 76.47,h 4.68,n 1.86;实测值:c 76.46,h 4.50,n 1.86。
[0190]
实施例26
[0191]
化合物238的制备
[0192]
s1.将合成实施例1中s3的2,4-二苯基-6-硼酸频哪醇酯吡啶(7.14g,20mmol)替换 为(2,6-二苯基嘧啶-4-基)硼酸(5.52g,20mmol),其他步骤同合成实施例1中s3,即可 得到6.73g 4-(8-溴噻蒽-2-基)-2,6-二苯基嘧啶,收率64%;
[0193]
s2.将合成实施例1中s4的(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.39g,10mmol)替换为(9
-ꢀ
苯基-9h-咔唑-3-基)硼酸(2.87g,10mmol),2-(7-溴噻蒽-2-基)-4,6-二苯基吡啶(5.24 g,10mmol)替换为4-(8-溴噻蒽-2-基)-2,6-二苯基嘧啶(6.72g,10mmol),其他合成步骤同 合成实施例1中的s4,即可得到5.02g 3-(7-(2,6-二苯基嘧啶-4-基)噻蒽-2-基-9-苯基-9h
-ꢀ
咔唑,收率73%;
[0194]
s3.在100ml三口瓶中,加入s2的9-(10-(7-(4-叔丁基苯基噻蒽-2-基)蒽-9-基)-9h
-ꢀ
咔唑(3.44g,5mmol)、二氯甲烷(30ml)、双氧水(5ml)、醋酸(15ml),升温至 70℃,反应2-6h,液相监测原料基本无剩余,降温停止反应。用硅胶漏斗过滤反应液, 过滤液水洗,分层,浓缩,即可得到3.64g目标化合物(238),收率97%;
[0195]
质谱仪maldi-tof-ms(m/z)=751.8764,理论分子量:751.8750;元素分析:理论 值c
46
h
29
n3(%):c 73.48,h 3.89,n 5.59;实测值:c 73.48,h 3.90,n 5.58。
[0196]
实施例27
[0197]
化合物246的制备
[0198]
s1.将合成实施例1中s3的2,4-二苯基-6-硼酸频哪醇酯吡啶(7.14g,20mmol)替换 为[1,1'-联苯]-3-基硼酸(3.96g,20mmol),其他步骤同合成实施例1中s3,即可得到6.00g 2-([1,1'-联苯]-3-基)-7-溴噻蒽,收率67%;
[0199]
s2.将合成实施例1中s4的(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.39g,10mmol)替换为 (3-(9h-咔唑-9-基)苯基)硼酸(2.87g,10mmol),2-(7-溴噻蒽-2-基)-4,6-二苯基吡啶(5.24 g,10mmol)替换为2-([1,1'-联苯]-3-基)-7-溴噻蒽(4.47g,10mmol),其他合成步骤同合成 实施例1中的s4,即可得到4.27g 9-(3-(7-([1,1'-联苯]-3-基)噻蒽-2-基)苯基)-9h-咔唑,收 率70%;
[0200]
s3.在100ml三口瓶中,加入s2的9-(3-(7-([1,1'-联苯]-3-基)噻蒽-2-基)苯基)-9h-咔 唑(3.05g,5mmol)、二氯甲烷(30ml)、双氧水(2.5ml)、醋酸(15ml),升温至 70℃,反应2-6h,液相监测原料基本无剩余,降温停止反应。用硅胶漏斗过滤反应液, 过滤液水洗,分层,浓缩,即可得到2.79g目标化合物(246),收率87%;
[0201]
质谱仪maldi-tof-ms(m/z)=641.8022,理论分子量:641.8030;元素分析:理论 值c
42
h
27
n(%):c 78.60,h 4.24,n 2.18;实测值:c 78.58,h 4.25,n 2.18。
[0202]
实施例28
[0203]
化合物267的制备
[0204]
s1.将合成实施例1中s3的2,4-二苯基-6-硼酸频哪醇酯吡啶(7.14g,20mmol)替换 为(10-苯基蒽-9-基)硼酸(5.96g,20mmol),其他步骤同合成实施例1中s3,即可得到 7.11g 2-溴-7-(10-苯基蒽-9-基)噻蒽,收率65%;
[0205]
s2.将合成实施例1中s4的(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.39g,10mmol)替换为(8
-ꢀ
苯基二苯并呋喃-2-基)硼酸(2.88g,10mmol),2-(7-溴噻蒽-2-基)-4,6-二苯基吡啶(5.24 g,10mmol)替换为2-溴-7-(10-苯基蒽-9-基)噻蒽(5.47g,10mmol),其他合成步骤同合成 实施例1中的s4,即可得到4.83g 2-苯基-8-(7-(10-苯基蒽-9-基)噻蒽-2-基)二苯并呋喃, 收率68%;
[0206]
s3.在100ml三口瓶中,加入s2的2-苯基-8-(7-(10-苯基蒽-9-基)噻蒽-2-基)二苯并 呋喃(3.55g,5mmol)、二氯甲烷(30ml)、双氧水(5ml)、醋酸(15ml),升温至 70℃,反应2-6h,液相监测原料基本无剩余,降温停止反应。用硅胶漏斗过滤反应液, 过滤液水洗,分层,浓缩,即可得到3.60g目标化合物(267),收率93%;
[0207]
质谱仪maldi-tof-ms(m/z)=774.9058,理论分子量:774.9050;元素分析:理论 值c
50
h
30
(%):c 77.50,h 3.90;实测值:c 77.50,h 3.92。
[0208]
实施例29
[0209]
化合物279的制备
[0210]
s1.将合成实施例1中s3的2,4-二苯基-6-硼酸频哪醇酯吡啶(7.14g,20mmol)替换 为(4-苯基萘-1-基)硼酸(4.96g,20mmol),其他步骤同合成实施例1中s3,即可得到6.76g 2-溴-7-(4-苯基萘-1-基)噻蒽,收率68%;
[0211]
s2.将合成实施例1中s4的(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.39g,10mmol)替换为(2
-ꢀ
苯基苯并噁唑-6-基)硼酸(2.39g,10mmol),2-(7-溴噻蒽-2-基)-4,6-二苯基吡啶(5.24 g,10mmol)替换为2-溴-7-(4-苯基萘-1-基)噻蒽(4.97g,10mmol),其他合成步骤同
合成 实施例1中的s4,即可得到3.79g 2-苯基-6-(7-(4-苯基萘-1-基)噻蒽-2-基)苯并噁唑,收率62%;
[0212]
s3.在100ml三口瓶中,加入s2的2-苯基-6-(7-(4-苯基萘-1-基)噻蒽-2-基)苯并噁唑 (3.06g,5mmol)、二氯甲烷(30ml)、双氧水(5ml)、醋酸(15ml),升温至70℃, 反应2-6h,液相监测原料基本无剩余,降温停止反应。用硅胶漏斗过滤反应液,过滤液 水洗,分层,浓缩,即可得到3.07g目标化合物(279),收率91%;
[0213]
质谱仪maldi-tof-ms(m/z)=675.7740,理论分子量:675.7730;元素分析:理论 值c
41
h
25
n(%):c 72.87,h 3.73,n 2.07;实测值:c 72.85,h 3.73,n 2.08。
[0214]
实施例30
[0215]
化合物284的制备
[0216]
s1.将合成实施例10中步骤s3的(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.37g,10mmol)替 换为(9-苯基-9h-咔唑-2-基)硼酸(2.87g,10mmol),其他合成过程同合成实施例步骤s2, 即可得到2.38g2,7-二(9-苯基-9h-咔唑-2-基)噻蒽,收率68%;
[0217]
s2.在50ml三口瓶中,加入s1的2,7-二(9-苯基-9h-咔唑-2-基)噻蒽(3.51g,5mmol)、 二氯甲烷(30ml)、双氧水(2.5ml)、醋酸(15ml),升温至70℃,反应2-6h,液相 监测原料基本无剩余,降温停止反应。用硅胶漏斗过滤反应液,过滤液水洗,分层,浓 缩,即可得到3.14g目标化合物(284),收率86%;
[0218]
质谱仪maldi-tof-ms(m/z)=730.9012,理论分子量:730.9000;元素分析:理论 值c
48
h
30
n2(%):c 78.88,h 4.14,n 3.83;实测值:c 78.88,h 4.16,n 3.82。
[0219]
实施例31
[0220]
化合物303的制备
[0221]
s1.将合成实施例10中步骤s3的(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.37g,10mmol)替 换为(3-(9h-咔唑-9-基)苯基)硼酸(2.87g,15mmol),其他合成过程同合成实施例步骤s3, 即可得到2.20g2,7-二(3-(9h-咔唑-9-基)苯基)噻蒽,收率63%;
[0222]
s2.在50ml三口瓶中,加入s1的2,7-二(3-(9h-咔唑-9-基)苯基)噻蒽(3.49g,5mmol)、 二氯甲烷(30ml)、双氧水(5ml)、醋酸(15ml),升温至70℃,反应2-6h,液相 监测原料基本无剩余,降温停止反应。用硅胶漏斗过滤反应液,过滤液水洗,分层,浓 缩,即可得到3.51g目标化合物(303),收率92%;
[0223]
质谱仪maldi-tof-ms(m/z)=762.8973,理论分子量:762.8980;元素分析:理论 值c
48
h
30
n2(%):c 75.57,h 3.96,n 3.67;实测值:c 75.57,h 3.95,n 3.69。
[0224]
实施例32
[0225]
化合物320的制备
[0226]
s1.将合成实施例1中s3的2,4-二苯基-6-硼酸频哪醇酯吡啶(7.14g,20mmol)替换 为(2-([1,1'-联苯]-3-基)-6-苯基嘧啶-4-基)硼酸(7.04g,20mmol),2,7-二溴噻蒽(9.35g,25 mmol)替换为2,7-二溴恶蒽(4.60g,25mmol),其他步骤同合成实施例1中s3,即可 得到6.95g2-([1,1'-联苯]-3-基)-4-(7-溴恶蒽-2-基)-6-苯基嘧啶,收率61%;
[0227]
s2.将合成实施例1中s4的(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.39g,10mmol)替换为(9
-ꢀ
苯基-9h-咔唑-3-基)硼酸(2.87g,10mmol),2-(7-溴噻蒽-2-基)-4,6-二苯基吡啶(5.24 g,10mmol)替换为2-([1,1'-联苯]-3-基)-4-(7-溴恶蒽-2-基)-6-苯基嘧啶(5.70g,
10mmol), 其他合成步骤同合成实施例1中的s4,即可得到5.40g目标化合物(320),收率74%。
[0228]
质谱仪maldi-tof-ms(m/z)=731.8562,理论分子量:731.8550;元素分析:理论 值c
52
h
33
n3(%):c 85.34,h 4.55,n 5.74;实测值:c 85.37,h 4.54,n 5.74。
[0229]
实施例33
[0230]
化合物323的制备
[0231]
s1.将合成实施例1中s3的2,4-二苯基-6-硼酸频哪醇酯吡啶(7.14g,20mmol)替换 为[1,1'-联苯]-3-基硼酸(3.96g,20mmol),2,7-二溴噻蒽(9.35g,25mmol)替换为2,7-二 溴恶蒽(4.60g,25mmol),其他步骤同合成实施例1中s3,即可得到5.56g2-([1,1'-联苯]-3
-ꢀ
基)-7-溴噻蒽,收率67%;
[0232]
s2.将合成实施例1中s4的(4-(9h-咔唑-9-基)萘-1-基)硼酸(3.39g,10mmol)替换为 咔唑(1.67g,10mmol),2-(7-溴噻蒽-2-基)-4,6-二苯基吡啶(5.24g,10mmol)替换为2-([1,1'
-ꢀ
联苯]-3-基)-7-溴噻蒽(4.15g,10mmol),其他合成步骤同合成实施例1中的s4,即可得 到3.76g目标化合物(323),收率73%。
[0233]
质谱仪maldi-tof-ms(m/z)=501.5863,理论分子量:501.5850;元素分析:理论值 c
36
h
23
n(%):c 86.21,h 4.62,n 2.79;实测值:c 86.20,h 4.62,n 2.80。
[0234]
实施例34
[0235]
化合物340的制备
[0236]
在100ml的三口瓶中,投入2,7-二溴恶蒽(3.42g,10mmol)、(9-苯基-9h-咔唑-3-基) 硼酸(6.32g,22mmol)、碳酸钾(2.76g,20mmol),按照2,7-二溴恶蒽的物质的量计,加入 2-3倍体积的甲苯、1-1.5倍体积的乙醇和1-1.5倍体积的水,在氮气氛围下,加入四(三 苯基膦)钯(0.03g,0.02mmol),升温至70-90℃反应6-18h,液相监测反应完成,冷却至室温, 反应液过滤,有机相浓缩后与滤饼一起拌样过硅胶柱,用10:1的石油醚与二氯甲烷的混 合液淋洗,淋洗液浓缩,即可得到4.00g目标化合物(340),收率60%。
[0237]
质谱仪maldi-tof-ms(m/z)=666.7790,理论分子量:666.7800;元素分析:理论 值c
48
h
30
n2(%):c 86.46,h 4.54,n 4.20;实测值:c 86.45,h 4.55,n 4.19。
[0238]
器件1-31的制备
[0239]
将100nm的氧化铟锡(ito)玻璃基板相继在清洗剂和去离子水中以超声波清洗1h, 之后先后用丙酮和异丙醇继续超声清洗15min,真空干燥2h(105℃),其后进行15min 的uv臭氧处理,将ito玻璃基板传送至真空蒸镀机。
[0240]
将三氧化钼(moo3)真空沉积在ito玻璃基板上至10nm的厚度形成空穴注入层。
[0241]
将n,n
’-
二苯基-n,n
’-
(1-萘基)-1,1
’-
联苯-4,4
’-
二胺(npb)真空沉积在空穴注入层上 至60nm的厚度形成空穴传输层。
[0242]
将4,4'-二(9-咔唑)联苯(cbp)(作为发光层主体材料)和三(2-苯基吡啶)合铱(ir (ppy)3)(作为发光层客体材料)以95:5的重量比共同真空沉积在空穴传输层上至 20nm的厚度形成发光层。
[0243]
将1,3,5-三(1-苯基-1h-苯并咪唑-2-基)苯(tpbi)真空沉积在发光层上至30nm的厚 度形成电子传输层材料。
[0244]
将氟化锂(lif)真空沉积在电子传输层上至1nm的厚度形成电子注入层。
[0245]
将镁银合金(mg/ag)真空沉积在电子注入层上至100nm的厚度形成阴极。
[0246]
分别采用化合物21、33、55、68、74、88、94、95、108、126、150、154、159、176、191、212、217、224、238、246、267、279、284、320、323以及下列结构式的对比物 1-3及alq3(8-羟基喹啉和铝)真空沉积在阴极上至60nm的厚度形成出光层材料,从而 完成绿光有机发光器件1-30的制备,并按照上述相同的器件制备方法,除了不蒸镀出光 层化合物(空白),制备无出光层的绿光有机电致发光器件31。对制备的发光器件1-31 进行性能检测。
[0247][0248]
具体检测数据见表1:
[0249]
表1有机电致发光器件性能表征
[0250]
[0251][0252]
检测结果表明,基于本发明的杂蒽衍生物在杂蒽基团的2,7位设计特定基团,从而 构建得到的本申请中的杂蒽衍生物表现出了优异的出光层材料特性。由表1可知,将本 申请的杂蒽衍生物作为出光层材料制备得到的有机电致发光器件,相比于同样结构的没 有出光层的有机电致发光器件以及同样结构利用传统的出光层材料,比如alq3,在电流 效率和外量子效率上具有显著的优势,增大幅度达到1.15倍以上;同时,本申请中的杂 蒽衍生物在杂蒽的2,7位修饰特定基团后,由于偏向于线型的空间构型,使得材料蒸镀 成膜后定向排列,具体如图1和图2,图1为基于本发明化合物94蒸镀得到的出光层膜 层的原子力显微镜成像图,图2为对比物2蒸镀得到的出光层膜层的原子力显微镜成像 图,基于本发明的化合物微观状态下的膜表面更为光滑,有利于作为出光层增强oled 器件光的耦合输出能力,使得基于本发明的杂蒽衍生物作为出光层制备的器件1-26,相 对于具有分支结构的对比物2作为出光层制备的器件28以及对比物3作为出光层制备的 器件29,在电流效率和外量子效率上具有显著的优势,增幅达到6%以上,即基于本发 明制备的器件具有较高的出光效率;此外,基于本发明的杂蒽衍生物在杂蒽的2,7位采 用不对称修饰,可强化非金属重
原子s在杂蒽衍生物的重原子效应和共轭离域程度,使 得杂蒽衍生物具有更高的玻璃化转变温度和高折射低吸收特性,诸如上表中公开的器件 1-23的玻璃化转变温度和折射率普遍优于器件24-26。基于本发明的2,7位特定基团修 饰的杂蒽衍生物,相比于对比物1-3以及现有技术中常用的出光层材料,具有显著优异 的出光特性,能更好的解决有机材料和基底间的折射率差异致使有机电致发光器件内全 反射造成的光的损耗,是一种理想的出光层材料。
[0253]
器件32-44的制备
[0254]
将器件实施例1中的发光层材料替换为95:5重量比的3-叔丁基-9,10-二(2-萘)蒽 (madn)(作为发光层主体材料)和4,4
’-
双(9-乙基-3-咔唑乙烯基)-1,1
’-
联苯(bczvbi) (作为发光层客体材料),出光层材料分别为化合物21、49、64、94、102、154、185、 201、303、340、对比物、alq3,其他制备过程同器件实施例1,制备得到蓝光器件32-43, 并按照上述相同的器件制备方法,除了不蒸镀出光层化合物(空白),制备无出光层的 有机电致发光器件44,对制备的发光器件进行性能检测。具体检测数据见表1:
[0255]
表2有机电致发光器件性能表征
[0256][0257]
检测结果表明,基于本发明的杂蒽衍生物作为出光层材料制备得到的有机电致发光 器件,相比于同样结构的没有出光层的有机电致发光器件以及同样结构利用传统的出光 层材料alq3,在电流效率和外量子效率上具有显著的优势,增大幅度达到1.13倍以上; 相对于具有分支结构的对比物2,在电流效率和外量子效率上具有显著的优势,增幅达到 2.7%以上。基于本发明的杂蒽衍生物在杂蒽的2,7位采用不对称修饰的杂蒽衍生物,相 比于对比物1-3以及现有技术中常用的出光层材料,具有显著优异的出光特性,能更好 的解
决有机材料和基底间的折射率差异致使有机电致发光器件内全反射造成的光的损 耗,同时,其不局限于某一种光色和电极之间的材料层结构,均表现出进一步提升电致 发光器件发光效率的特质,是一种理想的出光层材料。
[0258]
以上所述仅为本发明的较佳实施例,并不用以限制本发明,本领域的普通技术人员 可以理解:在不脱离本发明的原理和宗旨的情况下可以对这些实施例进行多种变化、修 改、替换和变型,本发明的范围由权利要求及其等同物限定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1