类蛇毒三肽的制备方法与流程
1.本发明涉及化工合成领域,特别是涉及类蛇毒三肽的制备方法。
背景技术:
2.类蛇毒三肽(h-β-ala-pro-dab-nh-bzl)是一种模拟蛇毒毒素waglerini活性的小肽,waglerini发现于temple viper毒蛇的毒液(tripidolaemus wagleri),为肌肉烟碱乙酰胆碱受体(nmachr)拮抗剂。h-β-ala-pro-dab-nh-bzl最早是由dsm荷兰皇家帝斯曼集团全资子公司pentapharm通过高科技生化科技,模仿有效分子蛋白(waglerin1)的片段合成得到,其化学结构与毒蛇血清相似,但是相比肉毒杆菌却更安全有效,故可安全应用到化妆品中,具有优秀的光滑肌肤、迅速祛皱性能。
3.目前常规合成类蛇毒三肽(h-β-ala-pro-dab-nh-bzl)的方法大多采用固相、液相相结合的方法,合成过程比较繁杂、成本高,为了在一定程度上简化工艺,研究者们开发了全液相合成类蛇毒三肽的方法,比如专利cn107936108a公开了如下合成路线:
[0004][0005]
该方法将boc-β-ala-oh、n-乙基-5-苯基异噁唑-3
’-
磺酸内盐和h-pro-ome-hcl反应得boc-β-ala-pro-ome,boc-β-ala-pro-ome与lioh反应得boc-β-ala-pro-oh;然后采用相同方法以boc-β-ala-pro-oh和h-dab(boc)-ome-hcl合成boc-β-ala-pro dab(boc)-oh;
再将boc-β-ala-pro-dab(boc)-oh、1-羟基苯丙三唑、n,n-二异丙基碳二亚胺和苄胺反应得boc-β-ala-pro-dab(boc)-nh-bzl,最后用三氟乙酸脱去boc保护基并经hplc分离纯化,得到纯度98%左右的类蛇毒三肽,总产率仅为38%左右,且合成路线较长,处理步骤繁琐,合成过程中许多试剂的用量较大。
[0006]
专利cn107857797a公开了另一种合成类蛇毒三肽的方法,合成线路如下:
[0007][0008]
该方法总产率也仅为30%左右,且仍需hplc分离纯化处理后才能得到纯度较高的类蛇毒三肽产品,合成步骤长,操作繁琐。
[0009]
由此可以看出,目前合成类蛇毒三肽的方法存在着产量低,合成工艺繁琐等问题,进而直接导致了实际生产中成本较高的问题。
[0010]
因此,开发工艺简便、产量高、成本低的类蛇毒三肽合成方法对类蛇毒三肽是生产及其在化妆品领域的应用具有重要意义。
技术实现要素:
[0011]
本发明主要解决的技术问题是提供一种类蛇毒三肽的制备方法,能够高效合成类蛇毒三肽,工艺简单,产率高。
[0012]
为解决上述技术问题,本发明采用的一个技术方案是:
[0013]
本发明提供boc-beta-ala-pro-dab(boc)-nhbn的制备方法,包括如下内容:由boc-beta-ala-pro-oh与h-dab(boc)-nhbn进行氨基酸脱水缩合反应得到:
[0014][0015]
进一步地,boc-beta-ala-pro-oh:h-dab(boc)-nhbn的摩尔用量比为1.0~1.5:1.0,优选1.1:1.0。
[0016]
进一步地,反应条件包括第一缩合剂;进一步地,所述第一缩合剂选自dic、dcc、edci中的一种或几种,优选dic;
[0017]
进一步地,所述第一缩合剂:h-dab(boc)-nhbn的摩尔用量比为1.0~1.5:1.0,优选1.2:1.0。
[0018]
进一步地,反应的溶剂为第一溶剂,所述第一溶剂选自dcm、etoac、thf中的一种或几种,优选dcm。
[0019]
进一步地,h-dab(boc)-nhbn:第一溶剂的质量用量比为1:15~30;进一步为1:20。
[0020]
进一步地,反应结束后将反应液分别用碱性水溶液、酸性水溶液洗、水洗涤,洗涤后固液分离,固形物用乙酸乙酯:石油醚质量比0.8~1.2:0.8~1.2的有机溶剂结晶纯化。
[0021]
所述结晶纯化是合成工艺中常用的纯化方法,即先通过升温等手段将固形物完全溶解在溶剂中,再通过降温等手段使其析出固体。
[0022]
所述碱性水溶液指呈碱性的水溶液,所述酸性水溶液指呈酸性的水溶液,不限定溶质,只要是不与产物boc-beta-ala-pro-dab(boc)-nhbn发生反应的碱性水溶液或酸性水溶液均适用。
[0023]
进一步地,所述碱性水溶液为质量分数为2~10%的碳酸钠水溶液,优选质量分数为5%的碳酸钠水溶液。
[0024]
进一步地,所述酸性水溶液为质量分数为2~10%的柠檬酸水溶液,优选质量分数为5%的柠檬酸水溶液。
[0025]
进一步地,所述有机溶剂的质量用量为h-dab(boc)-nhbn质量的15~25倍,进一步为20倍。
[0026]
在本发明中,结晶纯化是先将固形物和有机溶剂混合,升温至固形物完全溶解,再降温,析出固体。通常情况下,升温至60℃左右固形物可完全溶解。
[0027]
在本发明的具体实施例中,是将固形物和乙酸乙酯:石油醚质量比1:1的有机溶剂混合后,升温至固形物完全溶解,再降温,析出固体。
[0028]
进一步地,所述h-dab(boc)-nhbn是通过fmoc-dab(boc)-oh与苄胺进行氨基酸脱水缩合反应,再脱除fmoc基团得到:
[0029]
[0030]
氨基酸分子结合的方式是由一个氨基酸分子的羧基(—cooh)和另一个氨基酸分子的氨基(—nh2)结合连接,同时脱去一分子水,这种结合方式叫做氨基酸脱水缩合。
[0031]
进一步地,fmoc-dab(boc)-oh:苄胺的摩尔用量比为0.8~1.2:0.8~1.2,优选1:1。
[0032]
进一步地,fmoc-dab(boc)-oh与苄胺的氨基酸脱水缩合反应的条件包括第二缩合剂;进一步地,所述第二缩合剂选自dic、dcc、edci、hobt、cdi、hbtu、hatu中的一种或几种,优选dic和/或hobt,或选自edci;更优选dic和hobt,dic:hobt摩尔用量比为1:1。
[0033]
进一步地,所述第二缩合剂:fmoc-dab(boc)-oh的摩尔比用量为1.0~3.0:1.0,优选1.2~2.4:1.0。
[0034]
进一步地,fmoc-dab(boc)-oh与苄胺的氨基酸脱水缩合反应的溶剂为第二溶剂,所述第二溶剂选自dcm、thf、乙腈中的一种或几种,优选dcm和/或thf,更优选dcm。
[0035]
进一步地,fmoc-dab(boc)-oh:第二溶剂的质量用量比为1:5~15,优选1:10。
[0036]
脱除fmoc基团是常见的保护基团的脱除,本领域已知的脱除fmoc基团的方法均可应用于本发明。
[0037]
在本发明的具体实施方式中,是将fmoc-dab(boc)-nhbn、第三溶剂、六氢吡啶混合反应,所述溶剂选自甲醇、dcm、thf中的一种或几种,优选甲醇。
[0038]
进一步地,fmoc-dab(boc)-nhbn:第三溶剂的质量用量比为1.0:5~20,优选1.0:10。
[0039]
进一步地,fmoc-dab(boc)-nhbn:六氢吡啶的摩尔比用量为1.0:2~10,优选1.0:5。
[0040]
更进一步地,将反应后的体系移除溶剂后,将残余物与稀盐酸混合,再用dcm和/或乙酸乙酯萃取后,调节水相至碱性,再用乙酸乙酯萃取,将有机相移除溶剂后即得脱fmoc基团的产物h-dab(boc)-nhbn。
[0041]
在本发明的具体实施方式中,调节水相至碱性时,是用碳酸钠调节水相至ph为10。
[0042]
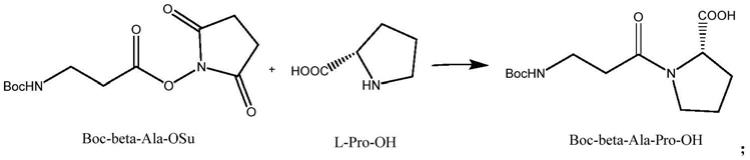
进一步地,所述boc-beta-ala-pro-oh是通过boc-beta-ala-osu与l-pro-oh发生如下反应得到:
[0043][0044]
进一步地,boc-beta-ala-osu:l-pro-oh的摩尔用量比为1.0:1.0~1.5,优选1;1.2;
[0045]
进一步地,boc-beta-ala-osu与l-pro-oh反应的条件包括碱;进一步地,所述碱选自nahco3、khco3、na2co3、k2co3中的一种或几种,优选nahco3;
[0046]
进一步地,boc-beta-ala-osu:碱的摩尔用量比为1.0:1.5~3,优选为1.0:2.0。
[0047]
进一步地,boc-beta-ala-osu与l-pro-oh反应的溶剂为水:有机溶剂摩尔用量比为0.8~1.2:0.8~1.2的混合溶剂,优选水:有机溶剂摩尔用量比为1:1的混合溶剂,所述有机溶剂选自四氢呋喃、乙腈、丙酮、甲醇、乙醇、dmf中的一种或几种,优选四氢呋喃、乙腈、丙
酮中的一种或几种,更优选四氢呋喃;
[0048]
更进一步地,boc-beta-ala-osu:混合溶剂的质量用量比为1:5~15,优选1:10。
[0049]
本发明还提供了一种制备类蛇毒三肽二盐酸盐的方法,通过boc-beta-ala-pro-dab(boc)-nhbn脱除boc基团得到;其中,boc-beta-ala-pro-dab(boc)-nhbn是通过前述任一种方法制备。
[0050]
进一步地,是将boc-beta-ala-pro-dab(boc)-nhbn与乙酸乙酯混合,再向溶液中分批次加入hcl的乙酸乙酯溶液,反应结束后固液分离得到固形物为类蛇毒三肽二盐酸盐。
[0051]
所述分批次加入,是指非一次性加入,可以是以一定的或不同的加料速度在一段时间内将原料加入,也可以是将需加入的原料分为若干份,在一定的时间内分若干次加入。
[0052]
进一步地,boc-beta-ala-pro-dab(boc)-nhbn:乙酸乙酯的质量用量为1:5~15,优选1:10;boc-beta-ala-pro-dab(boc)-nhbn:hcl的乙酸乙酯溶液的质量用量比为1:5~15,优选1:10。
[0053]
本发明还提供了类蛇毒三肽二盐酸盐在制备类蛇毒三肽相关产品中的用途,其中,所述类蛇毒三肽二盐酸盐是通过上述制备方法得到的。
[0054]
所述类蛇毒三肽相关产品,泛指含有类蛇毒三肽不同形式的化合物的产品,比如含有类蛇毒三肽、类蛇毒三肽不同形式的盐等,所述盐可以是盐酸盐、醋酸盐等。
[0055]
通过上述方法制得类蛇毒三肽二盐酸盐后,可利用现有的常规方法对其进行处理后得到类蛇毒三肽多种形式的化合物,比如,采用不同的酸处理转换为其它的盐(如类蛇毒三肽二醋酸盐)通过碱化处理可得类蛇毒三肽后,作为产品;或,将类蛇毒三肽不同形式的盐和/或类蛇毒三肽再进一步加工制备得到产品,如护肤品、药品等。只要在产品制备过程中,采用了本发明方法制备了类蛇毒三肽二盐酸盐,均属于本发明的保护范围。
[0056]
本发明的有益效果是:
[0057]
(1)本发明方法采用新的合成路线,无需hplc分离纯化即可得到纯度大于98%的产品,工艺简单,操作方便,且试剂用量少,产率高。
[0058]
(2)本发明方法合成工艺高效便捷,可大大降低生产成本,更适合实际生产应用,适合大规模工业化生产,值得推广。
[0059]
下面的缩写具有如下所示的意义:
[0060]
dcc:二环己基碳二亚胺;
[0061]
dic:二异丙基碳二亚胺;
[0062]
edci:1-(3-二甲胺基丙基)-3-乙基碳二亚胺;
[0063]
hatu:2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸盐;
[0064]
hbtu:o-苯并三氮唑-四甲基脲六氟磷酸酯;
[0065]
hobt:1-羟基苯并三唑;
[0066]
cdi:n,n'-羰基二咪唑;
[0067]
dcm:二氯甲烷;
[0068]
dmf:n,n-二甲基甲酰胺;
[0069]
thf:四氢呋喃。
具体实施方式
[0070]
下面对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0071]
本发明实施例中的收率均指摩尔收率。
[0072]
实施例中“eq”是指当量,用来表示以某一个试剂为基础(即1eq),其它试剂摩尔用量的倍数,“n倍重量份”是指质量用量为1eq试剂质量的n倍。
[0073]
例如,“向反应瓶中加入fmoc-dab(boc)-oh(1eq),二氯甲烷(10倍重量份),搅拌后溶解依次加入dic(1.2eq)”,是指dic的摩尔用量是fmoc-dab(boc)-oh的1.2倍,若fmoc-dab(boc)-oh的用量为1mol,则dic的用量为1.2mol;二氯甲烷的质量用量是fmoc-dab(boc)-oh的10倍,若fmoc-dab(boc)-oh的用量为1kg,则二氯甲烷的用量为10kg;其余情况同理。
[0074]
本发明实施例记载的方案合成规模均在10kg/批次,即每批次制得的类蛇毒三肽二盐酸盐质量为10kg左右。
[0075]
合成路线:
[0076]
步骤1:boc-beta-ala-pro-oh的合成:
[0077][0078]
步骤2:h-dab(boc)-nhbn的合成:
[0079][0080]
步骤3:类蛇毒肽的合成:
[0081]
[0082]
下面将通过不同的实施例说明本发明制备方法的具体效果:
[0083]
实施例1
[0084]
(1)boc-beta-ala-pro-oh的合成
[0085]
向反应瓶中加入l-pro-oh(1.2eq)、水(5倍重量份)、nahco3(2eq),四氢呋喃(5倍重量份),搅拌溶解后,室温加入boc-beta-ala-osu(1eq),然后在室温搅拌直到tlc监控反应完全。将反应液减压浓缩,稀盐酸调ph为3-4,乙酸乙酯(3x5倍重量份)萃取,无水na2so4干燥,过滤浓缩得红棕色油状物boc-beta-ala-pro-oh(收率80%)。
[0086]
boc-β-ala-pro-oh的1h nmr(d6-dmso):1.38(9h,s,3xch3);1.80-1.95(2h,m,ch2);2.08-2.20(1h,m,chh);2.33-2.48(3h,m,ch2,chh);3.10-3.15(2h,m,ch2);3.43-3.55(2h,m,ch2);4.21(1h,dd,j 8.0,4.0hz,ch);6.64(1h,m,nh);12.5(1h,br,oh)。
[0087]
(2)h-dab(boc)-nhbn的合成
[0088]
向反应瓶中加入fmoc-dab(boc)-oh(1eq),二氯甲烷(10倍重量份,以fmoc-dab(boc)-oh为基准),搅拌后溶解依次加入dic(1.2eq),hobt(1.2eq),室温搅拌30分钟后再加入苄胺(1eq);室温反应直到tlc监控反应完全。反应液先后用稀盐酸,水,饱和盐水洗涤,无水硫酸钠干燥,过滤后浓缩得fmoc-dab(boc)-nhbn,未经纯化直接投下一步反应(少量分析纯样品通过快速硅胶柱获得用于结构鉴定)。
[0089]
fmoc-dab(boc)-nhbn的1h nmr(d6-dmso):1.38(9h,s,3xch3);1.66-1.87(2h,m,ch2);2.93-3.02(2h,m,ch2);3.35(2h,s,ch2);4.03-4.08(1h,m,ch);4.21-4.31(5h,m,ch,2xch2);6.75(1h,br,nh);7.19-7.35(7h,m,ch);7.40-7.44(2h,m,2xch);7.60(1h,br,nh);7.74-7.75(2h,m,2xch);7.81-7.91(2h,m,2xch);8.37-8.39(1h,br,nh)。
[0090]
将上步得到得fmoc-dab(boc)-nhbn溶于甲醇(10倍重量份),然后加入六氢吡啶(5eq),室温反应完全后,浓缩,残余物加入到稀盐酸中,然后用二氯甲烷萃取杂质后,水相再用碳酸钠调节ph到10,再用乙酸乙酯萃取,无水硫酸钠干燥,浓缩得产品h-dab(boc)-nhboc(纯度大于95%,两步反应收率70%)。
[0091]
h-l-dab(boc)-nhbn的1h nmr(d6-dmso):1.38-1.46(10h,m,3xch3,chh);1.70-1.79(1h,m,chh);1.92(2h,br,nh2);3.00-3.05(2h,m,ch2);3.15-3.19(1h,m,ch);4.28-4.30(2h,m,ch2);6.78(1h,br,nh);7.24-7.34(5h,m,5xch);8.37-8.39(1h,br,nh)。
[0092]
(3)类蛇毒肽的合成
[0093]
向反应瓶中加入二氯甲烷(20倍重量份),boc-beta-ala-pro-oh(1.1eq),搅拌溶解后加入dic(1.2eq),搅拌30分钟后加入h-dab(boc)-nhbn(1.0eq),室温搅拌反应,tlc监控反应完成后,分别用5%碳酸钠,5%的柠檬酸水溶液,水洗,无水硫酸钠干燥,过滤浓缩后的残留物用乙酸乙酯/石油醚质量比1:1的混合溶剂(20倍重量份)结晶得到boc-beta-ala-pro-dab(boc)-nhbn(收率88%,纯度大于98%)。
[0094]
boc-beta-ala-pro-dab(boc)-nhbn的1h nmr(d6-dmso):1.30-1.35(18h,m,6xch3);1.60-2.20(6h,m,3xch2);2.45-2.50(2h,m,ch2);2.85-2.95(2h,m,ch2);3.10-3.20(2h,m,ch2);3.40-3.60(2h,m,ch2),4.20-4.40(4h,m,2xch,ch2);6.50-6.75(2h,m,2x nh),7.24-7.34(5h,m,5xch);7.8(1m,br,nh),8.20(1h,br,nh)。
[0095]
将上步得到得的boc-beta-ala-pro-dab(boc)-nhbn(1eq)溶解在乙酸乙酯(10倍重量份)中,再滴加hcl/乙酸乙酯溶液(4mol/l,10倍重量份),tlc监控反应完全后,反应液
中析出的固体过滤,淋洗(5倍重量份乙酸乙酯),干燥后得到类蛇毒三肽的二盐酸盐(收率95%,纯度大于98%)。
[0096]1h nmr(d2o):1.70-1.80(1h,m,chh);1.81-1.98(2h,m,ch2);2.01-2.30(3h,m,chh,ch2);2.73-2.78(2h,m,ch2);2.90-3.35(4h,m,2xch2);3.48-3.6(2h,m,,ch2);4.30-4.40(4h,m,2xch2)7.20-7.45(5h,m,5x ch)。
[0097]
实施例2
[0098]
(1)boc-beta-ala-pro-oh的合成
[0099]
向反应瓶中加入l-pro-oh(1.2eq)、水(5倍重量份)、nahco3(2eq),乙腈(5倍重量份),搅拌溶解后,室温加入boc-beta-ala-osu(1eq,已知化合物),然后在室温搅拌直到tlc监控反应完全。将反应液减压浓缩,稀盐酸调ph为3-4,乙酸乙酯(3x5倍重量份)萃取,无水na2so4干燥,过滤浓缩得红棕色油状物boc-beta-ala-pro-oh(收率75%)。
[0100]
(2)h-dab(boc)-nhbn的合成
[0101]
向反应瓶中加入fmoc-dab(boc)-oh(1eq),四氢呋喃(10倍重量份,以fmoc-dab(boc)-oh为基准),搅拌后溶解依次加入dic(1.2eq),hobt(1.2eq),室温搅拌30分钟后再加入苄胺(1eq);室温反应直到tlc监控反应完全。反应液先后用稀盐酸,水,饱和盐水洗涤,乙酸乙酯萃取,无水硫酸钠干燥,过滤后浓缩得fmoc-dab(boc)-nhbn,未经纯化直接投下一步反应。
[0102]
将上步得到得fmoc-dab(boc)-nhbn溶于二氯甲烷(10eq),然后加入六氢吡啶(5eq),室温反应完全后,浓缩,残余物加入到稀盐酸中,然后用二氯甲烷萃取杂质后,水相再用碳酸钠调节ph到10,再用乙酸乙酯萃取,无水硫酸钠干燥,浓缩得产品h-dab(boc)-nhboc(纯度大于95%,两步反应收率65%)。
[0103]
(3)类蛇毒肽的合成
[0104]
向反应瓶中加入二氯甲烷(20倍重量份),boc-beta-ala-pro-oh(1.1eq),搅拌溶解后加入dic(1.2eq),搅拌30分钟后加入h-dab(boc)-nhbn(1.0eq),室温搅拌反应,tlc监控反应完成后,分别用5%碳酸钠,5%的柠檬酸水溶液,水洗,无水硫酸钠干燥,过滤浓缩后的残留物用乙酸乙酯/石油醚质量比1:1的混合溶剂(20倍重量份)结晶得到boc-beta-ala-pro-dab(boc)-nhbn(收率89%,纯度大于98%)。
[0105]
将上步得到得的boc-beta-ala-pro-dab(boc)-nhbn(1eq)溶解在乙酸乙酯(10倍重量份)中,再滴加hcl/乙酸乙酯溶液(4mol/l,10倍重量份),tlc监控反应完全后,反应液中析出的固体过滤,淋洗(5倍重量份乙酸乙酯),干燥后得到类蛇毒三肽的二盐酸盐(收率93%,纯度大于98%)。
[0106]
实施例3
[0107]
(1)boc-beta-ala-pro-oh的合成
[0108]
向反应瓶中加入l-pro-oh(1.2eq)、水(5倍重量份)、nahco3(2eq),丙酮(5倍重量份),搅拌溶解后,室温加入boc-beta-ala-osu(1eq,),然后在室温搅拌直到tlc监控反应完全。将反应液减压浓缩,稀盐酸调ph为3-4,乙酸乙酯(3x5倍重量份)萃取,无水na2so4干燥,过滤浓缩得红棕色油状物boc-beta-ala-pro-oh(收率78%。
[0109]
(2)h-dab(boc)-nhbn的合成
[0110]
向反应瓶中加入fmoc-dab(boc)-oh(1eq),二氯甲烷(10倍重量份,以fmoc-dab
(boc)-oh为基准),搅拌后溶解依次加入edci(1.2eq),室温搅拌30分钟后再加入苄胺(1eq);室温反应直到tlc监控反应完全。反应液先后用稀盐酸,水,饱和盐水洗涤,无水硫酸钠干燥,过滤后浓缩得fmoc-dab(boc)-nhbn,未经纯化直接投下一步反应。
[0111]
将上步得到得fmoc-dab(boc)-nhbn溶于四氢呋喃(10倍重量份),然后加入六氢吡啶(5eq),室温反应完全后,浓缩,残余物加入到稀盐酸中,然后用乙酸乙酯萃取杂质后,水相再用碳酸钠调节ph到10,再用乙酸乙酯萃取,无水硫酸钠干燥,浓缩得产品h-dab(boc)-nhboc(纯度大于95%,两步反应收率68%)。
[0112]
(3)类蛇毒肽的合成
[0113]
向反应瓶中加入二氯甲烷(20倍重量份),boc-beta-ala-pro-oh(1.1eq),搅拌溶解后加入dic(1.2eq),搅拌30分钟后加入h-dab(boc)-nhbn(1.0eq),室温搅拌反应,tlc监控反应完成后,分别用5%碳酸钠,5%的柠檬酸水溶液,水洗,无水硫酸钠干燥,过滤浓缩后的残留物用乙酸乙酯/石油醚质量比为1:1的混合溶剂(20倍重量份)结晶得到boc-beta-ala-pro-dab(boc)-nhbn(收率88%,纯度大于98%)。
[0114]
将上步得到得的boc-beta-ala-pro-dab(boc)-nhbn(1eq)溶解在乙酸乙酯(10倍重量份)中,再滴加hcl/乙酸乙酯溶液(4mol/l,10倍重量份),tlc监控反应完全后,反应液中析出的固体过滤,淋洗(5倍重量份乙酸乙酯),干燥后得到类蛇毒三肽的二盐酸盐(收率95%,纯度大于98%)。
[0115]
尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1