一种Lupane三萜系衍生物晶型及其制备方法与流程
一种lupane三萜系衍生物晶型及其制备方法
技术领域
1.本发明涉及药物晶型技术领域,具体涉及一种lupane三萜系衍生物晶型及其制备方法。
背景技术:
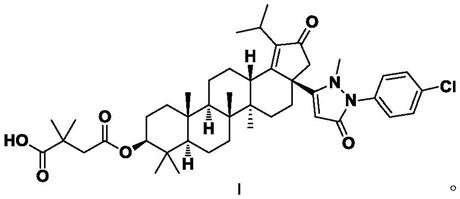
2.lupane三萜系衍生物是由江西青峰药业有限公司研究开发的用于抗hiv感染的药物,属于第二代hiv-1病毒成熟抑制剂。cn103242413a中公开了lupane三萜系衍生物4-(((3ar,5ar,5br,7ar,9s,11ar,11br,13as)-3a-(1-(4-氯苯基)-2-甲基-5-氧代-2,5-二氢-1h-吡唑-3-基)-1-异丙基-5a,5b,8,8,11a-五甲基-2氧代-3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a-十八烷氢-2h-环戊烷并-9基氧基)-2,2-二甲基-4-氧代-丁酸(式i所示),其对hiv病毒具有良好的活性。
[0003][0004]
目前,现有技术中尚未有关于上述式ⅰ结构lupane三萜系衍生物的晶型的报道,同一药物不同晶型在外观、溶解度、稳定性等方面可能有显著不同。因此,对式ⅰ结构的lupane三萜系衍生物进行研究开发合适的新晶型具有重要的意义。
技术实现要素:
[0005]
本发明的目的是提供一种式ⅰ结构lupane三萜系衍生物的新晶型及其制备方法,该晶型具有良好的固态稳定性和液态稳定性,其制备方法适合工业化规模生产。
[0006]
本发明的第一个方面,提供式ⅰ结构lupane三萜系衍生物的晶型a;
[0007][0008]
使用cu-kα辐射,其x射线粉末衍射图谱在以下2θ角度具有特征峰:6.61
°±
0.2
°
,8.84
°±
0.2
°
,13.20
°±
0.2
°
,13.90
°±
0.2
°
,14.64
±
0.2
°
,16.03
±
0.2
°
。
[0009]
优选地,其x射线粉末衍射图谱还在以下2θ角度具有一个或多个特征峰:12.68
°±
0.2
°
,15.29
°±
0.2
°
,18.71
°±
0.2
°
,20.22
°±
0.2
°
,20.97
°±
0.2
°
,21.85
°±
0.2
°
。
[0010]
优选地,其x射线粉末衍射图谱还在以下2θ角度具有一个或多个特征峰:10.57
°±
0.2
°
,12.26
°±
0.2
°
,17.23
°±
0.2
°
,17.73
°±
0.2
°
,22.18
°±
0.2
°
,22.89
°±
0.2
°
,26.57
°±
0.2
°
,29.52
°±
0.2
°
,35.50
°±
0.2
°
。
[0011]
优选地,其x射线粉末衍射图谱在以下2θ角度具有特征峰:
[0012]
[0013]
[0014][0015]
优选地,其x射线粉末衍射图基本上如图1所示。
[0016]
优选地,其差示扫描量热(dsc)图基本上如图2所示。
[0017]
优选地,其热重分析(tga)图基本上如图3所示。
[0018]
本发明中,“晶体”或“晶型”指的是被所示的x射线衍射图表征所证实的。本领域技术人员能够理解,其中的实验误差取决于仪器的条件,样品的准备和样品的纯度。特别是,本领域技术人员公知,x射线衍射图通常会随着仪器的条件而有所改变。特别需要指出的是,x射线衍射图的相对强度也可能随着实验条件的变化而变化,所以峰强度的顺序不能作为唯一或决定性因素。另外,峰角度的实验误差通常在5%或更少,这些角度的误差也应该被考虑进去,通常允许有
±
0.2
°
的误差;优选
±
0.1
°
的误差。另外,由于样品高度等实验因素的影响,会造成峰角度的整体偏移,通常允许一定的偏移。因而,本领域技术人员可以理解的是,任何具有本发明图谱的特征峰相同或相似的晶型均属于本发明的范畴之内。
[0019]
本发明所述的“晶体”或“晶型”是纯的、单一的,基本没有混合任何其它晶型。本发明中,“基本没有”当用来指新晶型时,指这个晶型含有少于20%(重量)的其他晶型,尤其指少于10%(重量)的其他晶型,更指少于5%(重量)的其他晶型,更指少于1%(重量)的其它品型。
[0020]
本发明的第二个方面,提供式ⅰ结构lupane三萜系衍生物晶型a的制备方法,该方法包含下述步骤:
[0021]
将式ⅰ结构lupane三萜系衍生物加入到结晶溶剂中,搅拌升温溶解后,再降温、析晶、过滤和干燥即得;
[0022]
所述结晶溶剂为乙腈;
[0023]
所述结晶溶剂与式ⅰ结构的lupane三萜系衍生物的体积质量比可以为8.0ml/g~20.0ml/g,优选地为10.0ml/g~15.0ml/g;
[0024]
所述式ⅰ结构lupane三萜系衍生物溶解的温度可以为60℃~90℃,优选地为70℃~80℃;
[0025]
所述降温析晶的降温速率可以为0.05℃/min~0.5℃/min,优选地为0.1℃/min~
0.2℃/min;
[0026]
所述降温析晶的析晶温度可以为0℃~15℃,优选地为5℃~10℃;
[0027]
所述干燥的温度可以为30℃~65℃,优选地为35℃~50℃;
[0028]
所述干燥的时间可以为20h~40h,优选地为24h~30h。
[0029]
本发明所用试剂和原料如在具体实施方式部分无特殊说明则为市售可得。
[0030]
本发明的第三个方面,提供上述晶型a或上述制备方法制得的晶型a在用于制备式i的其他晶型或其盐方面的应用。
[0031]
本发明的第四个方面,提供一种药物组合物,包含有效剂量的上述晶型a或上述制备方法制得的晶型a,以及药学上可接受的辅料。
[0032]
本发明的第五个方面,提供包含有效剂量的上述晶型a或上述制备方法制得的晶型a在用于制备预防或治疗hiv感染药物中的应用。
[0033]
本发明的第六个方面,提供一种用于预防或治疗hiv感染的联合药物组合物,包含有效剂量的上述晶型a或上述制备方法制得的晶型a,以及至少另一种治疗药物;优选地,所述至少另一种治疗药物选自:核苷/核苷酸逆转录酶抑制剂,非核苷类逆转录酶抑制剂,蛋白裂解酶抑制剂,融合抑制剂,进入抑制剂,和/或整合酶抑制剂。
[0034]
优选地,所述hiv为hiv-1。
[0035]
本发明具有的有益效果为:
[0036]
1.本发明提供的式ⅰ结构lupane三萜系衍生物晶型a,固态和液态稳定性均良好,可适应更宽松的制造、贮存和运输的环境条件,其能够更好地对抗由于温度、湿度、光照等因素可能产生的成分含量不均匀、纯度下降等问题,降低由此带来的风险。
[0037]
2.本发明提供的式ⅰ结构lupane三萜系衍生物晶型a的制备方法,制备得到的晶型a纯度高且收率高,工业操作可行性大,利于大规模工业化生产。
[0038]
因此,本发明提供的式ⅰ结构lupane三萜系衍生物晶型a及其制备方法适合产业化,具有广阔的工业应用前景。
附图说明
[0039]
图1所表示的为实施例1的式ⅰ结构lupane三萜系衍生物晶型a的x射线粉末衍射图;
[0040]
图2所表示的为实施例1的式ⅰ结构lupane三萜系衍生物晶型a的差示扫描量热图;
[0041]
图3所表示的为实施例1的式ⅰ结构lupane三萜系衍生物晶型a的热重分析图。
具体实施方式
[0042]
为了更好的理解本发明的内容,下面结合具体实施例来做进一步的说明,但具体的实施方式并不是对本发明的内容所做的限制。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0043]
本发明所述的x射线粉末衍射图在panalytical empyrean x射线粉末衍射仪上采集,本发明所述的x射线粉末衍射的方法参数如下:
[0044]
x射线反射参数:cu,kα
[0045]
[0046]
kα2/kα1强度比例:0.50
[0047]
电压:45千伏特(kv)
[0048]
电流:40毫安培(ma)
[0049]
扫描范围:自3.0至40.0:
[0050]
本发明所述的差示扫描量热(dsc)图在ta q2000上采集,本发明所述的差示扫描热量热(dsc)的方法参数如下:
[0051]
扫描速率:10℃/min
[0052]
保护气体:氮气
[0053]
本发明所述的热重分析(tga)图在ta q5000上采集,本发明所述的差示热重分析(tga)的方法参数如下:
[0054]
扫描速率:10℃/min
[0055]
保护气体:氮气
[0056]
本发明实施例中所用的各种试剂如无特别说明均为商购获得。
[0057]
本发明实施例中的温度如无特别说明均为室温。
[0058]
本发明实施例中的混合溶剂中的组分的比例如无特别说明均为体积比。
[0059]
实施例1:式ⅰ结构lupane三萜系衍生物晶型a的制备
[0060]
将50g式ⅰ结构lupane三萜系衍生物(可参照cn103242413a实施例1-73中公开的制备化合物98-1的方法制备得到)加入到1000ml反应瓶中,加入500ml乙腈,搅拌升温至80℃溶解后,降温至5℃析出白色晶体(降温速率0.2℃/min),过滤后,50℃干燥24h,即45.8g白色粉末,收率为91.62%,hplc纯度为99.57%,所得固体送检xprd,为晶型a。
[0061]
其xrd图如图1所示;
[0062]
其dsc图如图2所示,显示存在两个吸热峰,分别位于43.22℃和199.85℃;
[0063]
其tga图如图3所示,显示在150℃之前失重约3.0%;
[0064]
实施例2:式ⅰ结构lupane三萜系衍生物晶型a的制备
[0065]
将50g式ⅰ结构lupane三萜系衍生物(可参照cn103242413a实施例1-73中公开的制备化合物98-1的方法制备得到)加入到1000ml反应瓶中,加入750ml乙腈,搅拌升温至70℃溶解后,降温至10℃析出白色晶体(降温速率0.1℃/min),过滤后,35℃干燥30h,即44.98g白色粉末,收率为89.97%,hplc纯度为99.61%,所得固体送检xprd,为晶型a。
[0066]
实施例3:式ⅰ结构lupane三萜系衍生物晶型a的制备
[0067]
将50g式ⅰ结构lupane三萜系衍生物(可参照cn103242413a实施例1-73中公开的制备化合物98-1的方法制备得到)加入到2000ml反应瓶中,加入1000ml乙腈,搅拌升温至90℃溶解后,降温至0℃析出白色晶体(降温速率0.5℃/min),过滤后,65℃干燥40h,即42.68g白色粉末,收率为85.37%,hplc纯度为99.72%,所得固体送检xprd,为晶型a。
[0068]
实施例4:式ⅰ结构lupane三萜系衍生物晶型a的制备
[0069]
将50g式ⅰ结构lupane三萜系衍生物(可参照cn103242413a实施例1-73中公开的制备化合物98-1的方法制备得到)加入到2000ml反应瓶中,加入400ml乙腈,搅拌升温至60℃溶解后,降温至15℃析出白色晶体(降温速率0.05℃/min),过滤后,30℃干燥20h,即46.13g白色粉末,收率为92.25%,hplc纯度为99.52%,所得固体送检xprd,为晶型a。
[0070]
实施例5:固体稳定性
[0071]
在影响因素实验条件下(高温60℃,光照4500lx
±
500lx,高湿rh75%),考察其固体稳定性,实验结果如下表:
[0072][0073]
在高温、光照和高湿条件下,晶型a均稳定性良好。
[0074]
实施例5:溶液稳定性
[0075]
将实施例1所得晶型a用0.5%sls溶液配制,在48h内峰面积和保留时间rsd值均小于2%,表明溶液在测定时间内水溶液均稳定,具体情况如下表:
[0076]
样品不避光8h避光48h晶型a稳定稳定
[0077]
上述实验证明,本发明的晶型a具有良好固态稳定性和液态稳定性,可适应更宽松的制造、贮存和运输的环境条件,其制剂能够更好地对抗由于温度、湿度、光照等因素可能产生的成分含量不均匀、纯度下降等问题,降低由此带来的风险。
[0078]
本发明提供的式ⅰ结构lupane三萜系衍生物晶型a的制备方法,制备得到的晶型a纯度高、且收率高,工业操作可行性大,利于大规模工业化生产。
[0079]
尽管本发明的实施方案已公开如上,但其并不仅仅限于说明书和实施方式中所列运用。它完全可以被适用于各种适合本发明的领域。对于熟悉本领域的人员而言,可容易地实现另外的修改。因此在不背离权利要求及等同范围所限定的一般概念下,本发明并不限于特定的细节和这里示出与描述的图例。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1