一类假白榄烷型二萜关键中间体及其衍生物的合成方法与流程
[0001]
本发明属于化合物的合成领域,特别涉及一类假白榄烷型二萜关键中间体及衍生物的合成方法。
背景技术:
[0002]
大戟科植物广布于全球,但主产于热带和亚热带地区;在中国,主产地为西南和台湾地区。大戟属(euphorbia)作为大戟科最大的属,大戟属植物在传统中药里主要用于治疗水肿、偏头痛、结核、除疣等症。研究表明,该属植物主要化学成分为二萜、三萜和黄酮,而以结构多样的二萜为主;目前大约有650个二萜天然产物被分离得,大多都具有显著的生物活性,比如逆转肿瘤多药耐药、抗肿瘤、抗增殖、抗菌、抗炎、抗氧化等。其中一些活性好而副作用小的已经直接用作药物或者作为药物前体进而在临床上使用,例如,巨大戟醇(ingenol) 于2012年被美国fda批准用作治疗光线性角化病;对树脂胶毒素(resiniferatoxin)的研究目前也正处于临床iii期阶段。
[0003]
根据结构类型,这些二萜天然产物可以分为多种亚型,如半日花烷型(labdane)、克罗登烷型(clerodane)、松香烷型(abietane)、贝壳杉烷型(kaurane)、假白榄烷型(jatrophane) 等等。其中假白榄烷型大环二萜尤为突出,在众多亚型中占据核心地位,从生源出发,它是其他亚型的母体和基础;从结构上来说,该类二萜具有一个新颖的双环[10.3.0]十五烷基本骨架,含有多个手性中心(2-10个)并伴随有多个连续手性中心和季碳中心,其中构建含多手性中心、高度官能化的环戊烷片段是合成此类分子的重点和难点。
[0004]
基于该类天然产物出众的生物活性(已有多篇jmc文章报道假白榄型二萜具有强大的 pgp抑制活性)、天然来源的稀缺性,加之新颖且富有合成挑战性的化学结构,关于其合成也引起了合成化学家的广泛关注。但由于该类化合物结构复杂,目前只有少数两个结构相对简单的天然产物分子(手性中心2-4个)完成了全合成。
[0005]
发明人在前期从大戟科植物红雀珊珊中分离得8个全新的假白榄烷型大环二萜,并对部分化合物进行结构修饰获得22个新衍生物,将所得系列二萜进行多药耐药性相关的活性测试,发现了迄今为止活性最强的天然pgp抑制剂(cn106431925a)。如何通过人工合成的方式得这些化合物,具有非常重要的意义。
技术实现要素:
[0006]
本发明的目的在于提供一类假白榄烷型二萜关键中间体及衍生物的合成方法。
[0007]
本发明所采取的技术方案是:
[0008]
本发明的第一个方面,提供:
[0009]
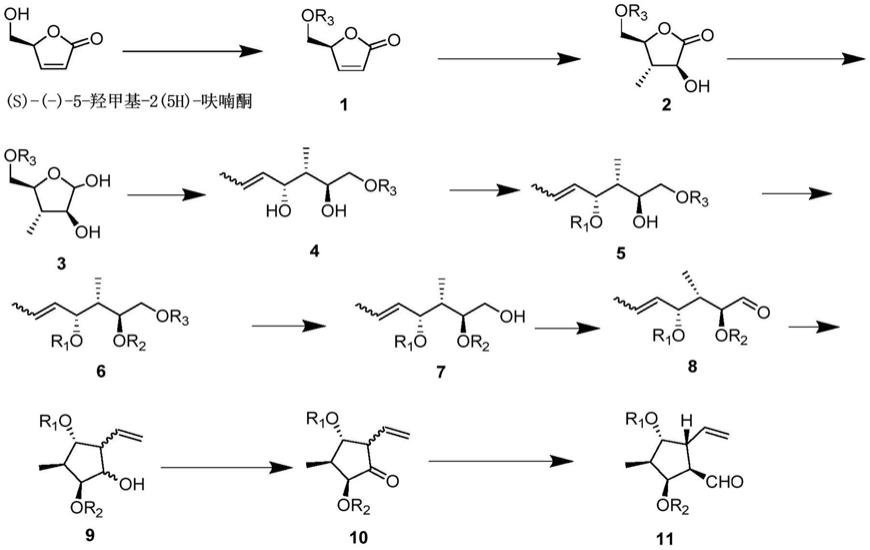
一类假白榄烷型二萜衍生物关键中间体,其结构式为:
[0010][0011]
式中,r1、r2独立为羟基保护基。
[0012]
在一些实例中,所述关键中间体的结构式为
[0013]
本发明的第二个方面,提供:
[0014]
一类假白榄烷型二萜衍生物关键中间体的合成方法,所述关键中间体如本发明的第一个方面所述,其合成路线如下:
[0015][0016]
式中,r1、r2、r3独立为羟基保护基;
[0017]
包括如下操作:
[0018]
以(s)-(-)-5-羟甲基-2(5h)-呋喃酮为起始原料,对其oh基进行保护,得化合物1;
[0019]
对化合物1进行甲基加成和羰基α位氧化反应得化合物2;
[0020]
化合物2经羰基还原反应得化合物3;
[0021]
化合物3经wittig反应得化合物4;
[0022]
将化合物4靠近烯键的羟基进行保护,得化合物5;
[0023]
将化合物5的羟基进行保护,得化合物6;
[0024]
脱去化合物6的保护基r3,得化合物7;
[0025]
将化合物7的羟基氧化得化合物8;
[0026]
化合物8经alder-ene反应得化合物9;
[0027]
化合物9的羟基经氧化反应得化合物10;
[0028]
化合物10经wittig反应得假白榄烷型二萜衍生物关键中间体化合物11。
[0029]
在一些实例中,各操作的反应条件独立如下:
[0030]
由化合物1合成化合物2的过程中,使用二甲基铜锂试剂与化合物1的有机溶剂溶液反应完全,萃取过滤之后加入冷冻干燥,干燥得到的粗产品溶解于有机溶剂中,加入双三甲基硅氨基锂和mooph,反应完全得到化合物2;
[0031]
化合物2与二异丁基氢化铝反应得到化合物3;
[0032]
乙基三苯基溴化磷与正丁基锂反应完全后,再加入化合物3的有机溶剂溶液,反应纯化得到化合物4;
[0033]
化合物7与过量戴斯马丁试剂反应,分离得到化合物8;
[0034]
化合物8的溶液升温至180~190℃反应完全,层析分离得到化合物9;
[0035]
将二甲基亚砜溶液和草酰氯混合,之后加入化合物9的溶液,反应完全后终止反应,分离得到化合物10;
[0036]
在甲氧基甲基三苯基氯化磷溶液中加入双三甲基硅氨基锂,之后加入化合物10的溶液反应,分离纯化得到化合物11。
[0037]
在一些实例中,各操作的反应条件独立如下:
[0038]
由化合物1合成化合物2的过程中,-20℃-30℃下,使用5-10个当量的二甲基铜锂试剂与化合物1的有机溶液反应完全,萃取过滤之后干燥,干燥得到的粗产品溶解于有机溶剂中,-50℃-60℃下,加入3-5个当量双三甲基硅氨基锂和2-3个当量的mooph,反应完全得到化合物2;
[0039]-50℃-78℃下,化合物2与2-3个当量的二异丁基氢化铝反应得到化合物3;
[0040]
乙基三苯基溴化磷与正丁基锂反应完全后,0℃-25℃下,再加入化合物3的有机溶剂溶液,反应纯化得到化合物4;
[0041]
化合物7与过量戴斯马丁试剂反应,分离得到化合物8;
[0042]
化合物8的溶液升温至180~190℃反应完全,柱层析分离得到化合物9;
[0043]-60℃-78℃下,将二甲基亚砜溶液和草酰氯混合,之后加入化合物9的溶液,反应完全后终止反应,分离得到化合物10;
[0044]
在甲氧基甲基三苯基氯化磷溶液中加入双三甲基硅氨基锂,0℃-25℃下,加入化合物10 的溶液反应,分离纯化得到化合物11。
[0045]
为了更好地脱保护,羟基保护基分别为不同的保护基,以便通过控制脱保护的条件,选择性脱除需要的保护基。在一些实例中,r1、r2、r3独立为tbdps、tbs、bz。
[0046]
在一些实例中,r1=tbs、r2=bz、r3=tbdps。
[0047]
本发明的第三个方面,提供:
[0048]
一类假白榄烷型二萜衍生物的合成方法,包括:
[0049]
磷酸酯片段的制备,其路线如下:
[0050][0051]
式中,r
11
、r5和r6为独立的羟基保护基;
[0052]
包括:
[0053]
异丁酸酯和3-甲基-2丁烯醛依次经aldol缩合、羟基脱水、羰基还原、羟基氧化反应得化合物17;
[0054]
化合物17和经evans不对称缩合并对反应得到的羟基进行保护,得到化合物18;
[0055]
化合物18醇解得到化合物19;
[0056]
化合物19经aldol反应得化合物20;
[0057]
假白榄烷型二萜关环,其合成路线如下:
[0058][0059]
包括:
[0060]
化合物11和化合物20经hwe反应得化合物21;
[0061]
化合物21经烯烃复分解反应得化合物22。
[0062]
在一些实例中,各操作的反应条件独立如下:
[0063]
在化合物12的溶液中加入二异丙基氨基锂反应,之后向反应液中滴入化合物13的溶液,反应完全后淬灭反应,分离得到化合物14;
[0064]
将化合物14溶解在有机溶剂中,加入对甲苯磺酸反应完全,分离得到化合物15;
[0065]
化合物15溶解在有机溶剂中,加入氢化铝锂反应得到化合物16;
[0066]
将草酰氯和二甲基亚砜溶液混合,加入化合物16反应,之后加入三乙胺继续反应,分离得到化合物17;
[0067]
向(n-乙酰基)-(4s)-异丙基-2-噁唑烷酮溶液中加入三氟甲磺酸二丁硼,混匀后加入化合物17的溶液,反应完全后加入pbs缓冲液和甲醇、双氧水和甲醇的混合溶液萃取,萃取液浓缩后溶解于有机溶剂中,加入羟基保护剂进行保护,得到化合物18;
[0068]
化合物18与甲醇钠在溶剂中醇解得到化合物19;
[0069]
化合物19、乙基磷酸二乙酯和正丁基锂在溶剂中反应得到化合物20;
[0070]
化合物11和化合物20在存在nah的溶剂中回流反应得到化合物21;
[0071]
化合物21在存在grubbs ii催化剂的溶剂中反应得到化合物22。
[0072]
在一些实例中,各操作的反应条件独立如下:
[0073]-50℃
--
78℃下,在化合物12的溶液中加入二异丙基氨基锂反应,之后向反应液中滴入化合物13的溶液,反应完全后淬灭反应,分离得到化合物14;
[0074]
将化合物14溶解在有机溶剂中,加入对1-3个当量的甲苯磺酸反应完全,分离得到化合物15;
[0075]
化合物15溶解在有机溶剂中,0℃-25℃下,加入2-3个当量氢化铝锂反应得到化合物 16;
[0076]
将草酰氯和二甲基亚砜溶液混合,-60℃
--
78℃下,加入化合物16反应,之后加入三乙胺继续反应,分离得到化合物17;
[0077]-50℃
--
78℃下,向(n-乙酰基)-(4s)-异丙基-2-噁唑烷酮溶液中加入三氟甲磺酸二丁硼,混匀后加入化合物17的溶液,反应完全后加入pbs缓冲液和甲醇、双氧水和甲醇的混合溶液萃取,萃取液浓缩后溶解于有机溶剂中,加入羟基保护剂进行保护,得到化合物18;
[0078]
化合物18与甲醇钠在溶剂中醇解得到化合物19;
[0079]
化合物19、乙基磷酸二乙酯和正丁基锂在溶剂中反应得到化合物20;
[0080]
化合物11和化合物20在nah的溶剂中回流反应得到化合物21;
[0081]
化合物21在催化量的grubbs ii催化剂的溶剂中反应得到化合物22。
[0082]
在一些实例中,所述假白榄烷型二萜衍生物选自:
[0083][0084]
为了更好地脱保护,羟基保护基分别为不同的保护基,以便通过控制脱保护的条件,选择性脱除需要的保护基。在一些实例中,r
11
、r5和r6独立为tbs和c2~c4的烷基;优选的,r
11
=tbs、r5和r6=相同或者不同的c2~c4的烷基;进一步的,r5和r6=乙基。
[0085]
本发明的有益效果是:
[0086]
本发明的一些实例,可以合成得到具备连续五个手性中心的一类假白榄烷型二萜关键中间体环戊烷片段。
[0087]
本发明的一些实例,该类发散性不对称合成路线简洁高效,相比于从植物中提取的低效率和不确定性,通过该路线可以大量并定向制备目标化合物,具有显著的实用性。
[0088]
本发明的一些实例,可以大量制备结构多样性的假白榄行二萜类衍生物,对于完善该类结构的构效关系,发现活性更好的先导化合物,发展新一代的基于pgp抑制剂的抗肿瘤药物具有良好的应用前景。
附图说明
[0089]
图1是化合物9的核磁共振氢谱;
[0090]
图2是化合物9的核磁共振碳谱;
[0091]
图3是化合物9的高分辨质谱;
[0092]
图4是化合物10的核磁共振氢谱;
[0093]
图5是化合物10的核磁共振碳谱;
[0094]
图6是化合物10的高分辨质谱。
具体实施方式
[0095]
通过对假白榄烷型大环二萜的结构进行分析,发现在假白榄烷型大环二萜的合成路线中,存在高度氧化态(含有多个氧原子)的环戊烷结构存在,为假白榄烷型大环二萜合成的关键中间体。
[0096]
该假白榄烷型大环二萜关键中间体存在如下难点:
[0097]
1)氧化态过高的结构往往会导致化合物不稳定。在这一个极其狭窄五元环空间中,需要依次将甲基、羟基、obz、otbs和烯丙基等各种基团添加在五元环中,且该五元环体系含有三个氧化度,这为该类结构的合成带来了极大的挑战。
[0098]
2)该环戊烷片段含有连续的5个手性中心,每个手性有两种可能构型,连续的5个手性意味着有32种可能的手性化合物,而产物只是其中的一个。每次引入新的手性中心的
过程中,若是控制不够好,要么全部变成另外的手性化合物导致路线失败,要么部分产生另外的手性损失产率,而无法进行下一步的反应。
[0099]
总之,多手性化合物特别是需要完美控制理想的连续5个手性中心的化合物的合成具有非常大的难度。
[0100]
该路线中部分反应如wittig反应、ene反应、迈克加成反应等可参考工具书jiejackli著. 有机人名反应机理及应用.北京:科学出版社,2011.07.,以及邢其毅等著.基础有机化学上第3版.北京:高等教育出版社,2005.06.或其他教科书或工具书。
[0101]
下面结合实例,进一步说明本发明的技术方案。
[0102]
中间体环戊烷片段的合成
[0103]
合成路线如下:
[0104][0105]
式中,r1=tbs、r2=bz、r3=tbdps。tbs:叔丁基二甲基硅烷;bz:苯甲酰基;tbdps:叔丁基二苯基硅烷。
[0106]
磷酸酯片段的制备,其路线如下:
0℃下,向烧瓶中缓慢加入甲基锂试剂(87.3ml,1.3m,113.5mmol)至澄清透明状。将反应液冷却至-20℃,将实施例一中得到的化合物1(11.4mmol)溶于无水乙醚(10ml),向反应液中缓慢加入。反应1h后,加入饱和氯化铵溶液(50ml)淬灭反应,用乙酸乙酯萃取三次,合并有机层,得粗产品。
[0116]
将粗品溶解在无水thf(30ml)中加入至三颈烧瓶中,置换n2后降温至-78℃。缓慢向反应液中滴入lihmds(24.49ml,1m in thf),反应20min后,经恒压滴液漏斗缓慢向其中滴入mooph的thf溶液。-78℃下反应2h,tlc监测至原料不再减少后,向反应液中加入nh4cl的饱和溶液淬灭反应。用etoac(90ml x 3)萃取,收集有机层,经柱层析纯化得到化合物2,无色油状液体。1h nmr(400mhz,cdcl3)δ7.67(m,4h),7.43(m,6h),4.09 (d,j=10.5hz,1h),4.04(m,1h),3.93(dd,j=12.0,2.5hz,1h),3.76(dd,j=12.0,4.0hz,1h), 2.47(m,1h),1.17(d,j=6.6hz,3h),1.06(s,9h).
13
c nmr(100mhz,cdcl3)δ176.6,135.63, 135.55,132.8,132.6,130.0,127.83,127.83,83.0,62.6,39.5,26.7,19.2,14.4.
[0117]
实施例三:化合物3的合成
[0118]
将实施例二中得到化合物2(2g,4.01mmol)溶解在无水dcm(10ml)中并加入至三颈烧瓶中,置换n2后降温至-78℃。缓慢向反应液中滴入dibal(4.81ml,1m in toluene),反应 1h后。向反应液中加入酒石酸钾钠的饱和溶液淬灭反应。室温下剧烈搅拌3h后,用etoac 萃取,收集有机层,经柱层析纯化得到化合物3,无色油状液体。1h nmr(400mhz,cdcl3) δ7.70(m,4h),7.44(m,6h),5.19(dd,j=7.6,3.6hz,1h),3.80(dd,j=11.1,2.7hz,1h),3.71(m, 1h),3.66(m,1h),3.53(dd,j=11.2,2.4hz,1h),2.18(m,1h),1.11(d,j=7.1hz,1h),1.09(s, 9h).
13
c nmr(100mhz,cdcl3)δ135.7,135.6,132.4,130.1,130.0,127.9,96.3,84.6,78.9,64.6, 38.5,26.9,26.8,19.2,15.5.hresims m/z 385.1843[m-h]-(calcd 385.1841).
[0119]
实施例四:化合物4的合成
[0120]
0℃,n2保护下向etpph3br(1.73g,4.66mmol)的无水thf(10ml)中加入n-buli (1.94ml,2.4m in toluene),反应10min。将实施例三中得到化合物3(450mg,0.258 mmol)的无水thf(5ml)溶液缓慢滴入后,0℃下反应2h。tlc监测至原料消失后,向反应液中加入饱和nh4cl溶液淬灭反应。收集有机层,减压浓缩后经柱层析纯化得到化合物 4,无色油状液体。y5-7-1-1n:z/e=5:1;e;1h nmr(400mhz,cdcl3)δ7.66(m,4h),7.42 (m,6h),5.59(m,2h),4.64(dd,j=7.7,2.5hz,1h),3.75(m,2h),3.61(m,1h),3.07(s,1h), 1.86(ddd,j=14.7,7.1,2.5hz,1h),1.64(d,j=5.4hz,3h),1.04(s,9h),0.76(d,j=7.1hz,3 h).
13
c nmr(100mhz,cdcl3)δ135.51,132.97,130.75,129.90,127.85,126.43,74.82,69.58, 66.46,40.22,26.86,19.25,13.44,11.77.
[0121]
y5-7-1-1n;z;1h nmr(400mhz,cdcl3)δ7.65(m,4h),7.41(m,6h),5.68(m,1h),4.64 (dd,j=15.3,6.5,1.4hz,1h),4.22(m,1h),3.73(m,2h),3.61(td,j=8.4,2.0hz,1h),1.81(m, 1h),1.72(d,j=6.3hz,3h),1.07(s,9h),0.75(d,j=7.1hz,3h).
13
c nmr(100mhz,cdcl3) δ135.53,133.01,132.98,131.11,129.90,127.82,127.14,74.85,66.46,39.79,26.87,19.26,17.81, 11.72.
[0122]
实施例五:化合物5的合成
[0123]
将实施例四中得到的的化合物4(450mg,1.17mmol)和咪唑(398.3mg,5.85mmol)溶
解在无水dcm(5ml)中。0℃下,向反应液中加入tbscl(529mg,3.51mmol)。10min 后,转至室温下反应2h。tlc监测至原料消失后,加入h2o淬灭反应。用etoac萃取,收集有机层,减压浓缩后经柱层析纯化得到化合物5,无色油状液体。5:1h nmr(400mhz,cdcl3) of a 2/1mixture of double bond isomer.δ7.70(m,4h
major
),7.70(m,4h
minor
),7.41(m,6h
major
), 7.41(m,6h
minor
),5.52(m,2h
major
),5.52(m,2h
minor
),4.84(dd,j=7.7,2.7hz,1h
major
),4.37(dd, j=6.4,2.7hz,1h
minor
),3.77(m,1h
major
),3.77(m,1h
minor
),3.69(m,1h
major
),3.69(m,1h
minor
), 3.64(m,2h
major
),3.64(m,2h
minor
),1.91(ddd,j=14.5,7.08,2.7hz,1h
major
),1.91(ddd,j=14.5, 7.08,2.7hz,1h
minor
),1.65(d,j=5.1hz,3h
major
),1.70(d,j=5.7hz,3h
minor
),1.07(s,9h
major
), 1.07(s,9h
minor
),0.90(s,9h
minor
),0.89(s,9h
major
),0.77(d,j=7.1hz,6h
major
),0.74(d,j=0.74 hz,3h
minor
),0.06(d,j=15.2hz,6h
major
),0.05(d,j=16.5hz,6h
minor
).
13
c nmr(100mhz, cdcl3)δ135.7
minor
,135.6
major
;135.65
minor 135.59
major
;133.4
minor
,132.2
major
;131.5
minor
,129.6
major
; 127.7
minor
,127.7
major
;126.6
minor
,124.4
major
;75.6
minor
,73.9
major
;70.0
minor
,70.0
major
;66.4
minor
,66.3
major
; 41.4
major
,41.1
minor
;26.8
major
,26.8
minor
;25.83
minor
,25.77
major
;19.3
major
,19.3
minor
;18.08
minor
,18.02
major
; 17.7
minor
,13.4
major
;-4.19
minor
,-4.33
major
;-5.14
minor
,-5.19
major
.
[0124]
实施例六:化合物6的合成
[0125]
将实施例五中得到的化合物5(300mg,0.6mmol)溶解在无水吡啶(5ml)中。0℃下,向反应液中加入bzcl(1.8mmol)。10min后,转至75℃下反应过夜。减压浓缩后经柱层析纯化得到化合物6。y5-7-1-3n:1h nmr(400mhz,cdcl3)of a 2.7/1mixture of double bond isomer.δ8.06(m,2h
major
),8.06(m,2h
minor
);7.60(m,3h
major
),7.60(m,3h
minor
);7.56(m,3h
major
), 7.56(m,3h
minor
);7.47(t,j=7.6hz,2h
major
),7.47(t,j=7.6hz,2h
minor
);7.37(t,j=7.4hz,1 h
major
),7.37(t,j=7.4hz,1h
minor
);7.29(m,2h
major
),7.29(m,2h
minor
);7.13(m,2h
major
),7.13 (m,2h
minor
);5.58(dd,j=7.6,1.7hz,1h
minor
),5.54(m,1h
major
);5.46(m,1h
major
),5.46(m,1 h
minor
);5.09(m,1h
major
),5.09(m,1h
minor
);4.72(dd,j=8.0,1.9hz,1h
major
),4.23(dd,j=5.8, 3.04hz,1h
minor
);4.00(dd,j=6.9,2.3hz,1h
minor
),3.97(dd,j=6.6,2.1hz,1h
major
);3.87(dd,j =4.4,3.8hz,1h
major
),3.89(dd,j=4.3,3.8hz,1h
minor
);2.30(m,1h
major
),2.24(m,1h
minor
);1.68 (dd,j=6.8,1.3hz,3h
major
),1.66(d,j=2.5hz,3h
minor
);1.02(m,3h
major
),0.99(m,3h
minor
);1.01 (s,9h
minor
),1.01(s,9h
major
);0.83(s,9h
minor
),0.83(s,9h
major
);-0.09(s,3h
minor
),-0.09(s,3h
major
);
ꢀ-
0.15(s,3h
major
),-0.15(s,3h
minor
).
13
c nmr(100mhz,cdcl3)δ165.7
major
,165.7
minor
;135.5
major
, 134.3
minor
,133.5
minor
,133.45
major
,133.34
minor
,133.31
major
,132.64
minor
,132.68
major
,130.91
minor
, 130.87
major
,129.62
minor
,129.58
major
,129.46
minor
,129.55
major
,128.25
minor
,128.3
major
,127.46
minor
, 127.6
major
,125.9
minor
,123.3
major
,73.6
minor
,67.5
major
,63.4
minor
,63.2
major
,40.8
minor
,40.5
major
,26.7
minor
, 26.7
major
,25.9
minor
,25.8
major
,19.2
minor
,19.3
major
,18.09
minor
,18.02
major
,17.7
minor
,17.6
major
,13.2
minor
, 13.2
major
,9.9
minor
,9.5
major
,-3.8
minor
,-4.2
major
,-5.2
minor
,-5.3
major
.
[0126]
实施例七:化合物7的合成
[0127]
将实施例七中得到化合物7(1.44mmol)溶于thf(10ml)中,0℃下加入70%hf
·
py继续反应10h。tlc监测至原料消失后,加入饱和的碳酸氢钠溶液淬灭反应。乙酸乙酯萃取
后,减压浓缩并经柱层析纯化后得到化合物7。7:1h nmr(400mhz,cdcl3)of a 2.5/1mixture of double bond isomer.δ8.07(m,2h
major
),8.07(m,2h
minor
);7.58(m,1h
major
),7.58(m,1h
minor
);7.45 (m,2h
major
),7.45(m,2h
minor
);5.59(m,1h minor),5.59(m,1h
major
);5.52(m,1h
minor
),5.52(m,1 h
major
);5.10(m,1h
minor
),5.10(m,1h
major
);4.20(d,j=6.6,3.3hz,1h
minor
),4.68(d,j=8.3,3.1 hz,1h
major
);3.99(m,1h
minor
),3.99(m,1h
major
);3.81(m,1h
minor
),3.81(m,1h
major
);2.65(t,j= 6.1hz,1h
minor
),2.48(t,j=5.8hz,1h
major
);2.12(m,1h
minor
),2.12(m,1h
major
);1.71(d,j=5.2 hz,3h minor),1.67(dd,j=6.6,1.2hz,3h
major
);0.99(d,j=7.3hz,3h
minor
),1.02(d,j=7.0hz, 3h
major
);0.87(s,9h
minor
),0.86(s,9h
major
);-0.03(s,3h
minor
),-0.06(s,3h
minor
),-0.04(s,3h
minor
),
ꢀ-
0.08(s,3h
major
).
13
c nmr(100mhz,cdcl3)δ166.70
major
,166.68
minor
;133.31
major
,133.03
minor
; 133.06
major
,132.8
minor
;130.27
minor
,130.25
major
;129.65
major
,129.61
minor
;128.41
major
,128.37
minor
; 126.53
minor
,123.93
major
;77.92
major
,77.78
minor
;74.88
minor
,68.2
major
;63.05
major
,63.00
minor
;41.4
minor
, 41.3
major
;25.84
minor
,25.79
major
;18.10
minor
,18.04
major
;17.6
minor
,13.31
major
;11.2
minor
,10.4
major
;-4.0
minor
,
ꢀ-
4.2
major
;-5.1
minor
,-5.3
major
.
[0128]
实施例八:化合物8的合成
[0129]
将实施例七中得到的化合物7(0.53mmol)溶于dcm(10ml)中,室温下加入dmp(1.6 mmol)反应3h。tlc监测至原料消失后,加入饱和的碳酸氢钠溶液淬灭反应。乙酸乙酯萃取后,减压浓缩并经柱层析纯化后得到化合物8。1h nmr(400mhz,cdcl3)δ9.65(d,j=0.9 hz,1h
major
),9.63(d,j=0.8hz,1h
minor
);8.09(m,2h
major
),8.09(m,2h
minor
);7.61(m,1h
major
), 7.61(m,1h
minor
);7.48(m,2h
major
);5.57(m,2h
major
),5.57(m,2h
minor
);7.48(m,2h
minor
);5.17 (dd,j=5.5,0.8hz,1h
major
),5.17(dd,j=5.1,0.9hz,1h minor
);4.14(dd,j=8.2,5.0hz,1h
major
), 4.27(dd,j=6.8,5.1hz,1h
minor
);2.44(m,1h
major
),2.44(m,1h
minor
);1.74(d,j=5.6hz,3h
minor
), 1.70(dd,j=6.5,1.2hz,3h
major
);1.11(d,j=10.3hz,3h
major
),1.11(d,j=10.3hz,3h
minor
);0.87 (s,9h
minor
),0.86(s,9h
major
);0.0(s,3h
major
),-0.01(s,3h
minor
);-0.01(s,3h
major
),-0.02(s,3h
minor
). 13
c nmr(100mhz,cdcl3)δ197.70
major
,197.62
minor
;166.04
major
,166.04
minor
;135.91
minor
,133.42
major
; 132.63
major
,132.58
minor
;129.80
minor
,129.77
major
,129.55
major
,129.55
minor
;129.34
minor
,129.30
major
; 128.53
major
,128.48
minor
;128.30
major
,128.30
minor
;127.39
major
,127.76
minor
;80.62major,80.51minor; 75.01
minor
,68.65
major
;42.23
major
,42.23
minor
;25.81minor,25.75major;18.12
minor
,18.07
major
;17.64
minor
, 13.53
major
;12.26
minor
,11.80
major
;-4.12
minor
,-4.36
major
;-5.00
minor
,-5.12
major
.
[0130]
实施例九:化合物9合成
[0131]
将实施例八中得到的化合物8(0.21mmol)溶于甲苯(7ml)中,加入至耐压管中,升温至 180-190℃下反应2d-3d。tlc检测至原料不再减少后,减压蒸馏再经柱层析纯化得到化合物9。9:1h nmr(400mhz,cdcl3)δ8.02(m,2h),7.57(m,1h),7.45(t,j=7.7hz,2h),6.03 (ddd,j=16.9,10.8,8.9hz,1h),5.42(d,j=7.7,2.2hz,1h),5.29(s,1h),5.25(m,1h),4.14(t,j =6.1hz,1h),3.95(t,j=3.5hz,1h),3.22(d,1h),2.83(m,1h),2.64(m,1h),1.04(d,j=7.5hz, 3h),0.90(s,9h),0.10(s,3h),0.07(s,3h);
13
c nmr(100mhz,cdcl3)δ166.2,133.4,133.1, 130.0,129.6,128.4,118.7,82.6,82.2,79.8,50.8,44.7,25.8,18.0,12.7,-4.7,-4.8。化合物9的核磁共振氢谱和碳谱以及高分辨质谱分别见图1~3。
3h,tlc监测至原料消失后向反应液中加入1%hcl淬灭反应。萃取后经柱层析分离得到化合物16。
[0142]
实施例十五:化合物17的合成
[0143]
取草酰氯(0.63ml,7.42mmol)溶解在二氯甲烷(5ml)中,n2保护并冷却至-78℃下。向反应液中缓慢滴入二甲基亚砜(0.8ml,11.2mmol)的二氯甲烷(5ml)溶液,反应30min。将实施例十四中得到的化合物16(260mg,1.85mmol)的二氯甲烷溶液(10ml)缓慢滴入反应液中,继续反应1h后,加入20个当量的三乙胺(5.14ml,37.1mmol)。转移至室温下搅拌10min后,加水萃取。减压蒸馏后经柱层析分离得到化合物17。1h nmr(400mhz,cdcl3) δ9.2(s,1h),6.02(d,j=17.2hz,1h),5.71(d,j=16.2hz,1h),4.86(m,1h),4.53(m,1h), 1.52(s,3h),1.35(s,6h);
13
c nmr(100mhz,cdcl3)δ198.2,136.5,131.4,129.6,112.9,42.6, 33.4,22.1,18.2.
[0144]
实施例十六:化合物18的合成
[0145]
取(n-乙酰基)-(4s)-异丙基-2-噁唑烷酮(68.1mg,0.4mmol)溶解在二氯甲烷(1ml) 溶液中,n2保护,冰浴下降温至0℃后,向反应液中加入1.2个当量的三氟甲磺酸二丁硼 (0.43ml,0.43mmol),反应20min。随后,向反应液中加入1.5个当量的三乙胺(0.067ml, 0.49mmol)。将反应液降温至-78℃后,取实施例十五中得到化合物17(50mg,0.36mmol) 的二氯甲烷(2ml)溶液缓慢滴入反应液中。将反应液移至室温下反应12h后,冰浴下向反应液中加入pbs缓冲液和甲醇淬灭反应,随后加入30%的h2o2的甲醇溶液,室温下搅拌2 h。反应液经乙酸乙酯萃取后减压浓缩得粗产品。将粗产品溶解在二氯甲烷溶液中后,降温至 0℃,向反应液中加入3个当量的咪唑以及1.5个当量的叔丁基二甲基氯硅烷。将反应液移至室温下反应1h后,加水淬灭,乙酸乙酯萃取后经柱层析分离得到化合物18。1h nmr(400 mhz,cdcl3)δ5.89(d,j=10.5hz,1h),5.60(d,j=11.5hz,1h),4.72(m,1h),4.32(ddd,j= 8.3,8.3,3.4hz,1h),4.12(dd,j=12.6,5.7hz,1h),4.02(dd,j=12.5,4.5hz,1h),3.60(t,j= 3.5hz,1h),2.56(m,2h),2.62(dd,j=10.5,4.6hz,1h),1.65(s,3h),0.98(s,9h),0.94(s,6 h),0.91(s,3h),0.89(s,3h);
13
c nmr(100mhz,cdcl3)δ166.2,155.2,135.3,128.6,121.2, 120.1,112.6,73.2,62.3,60.5,42.6,36.5,28.6,23.5,21.1,15.6,,-4.5,-3.6.
[0146]
实施例十七:化合物19的合成
[0147]
将实施例十六中得到的化合物18(100mg,0.23mmol)溶解在二氯甲烷中,降温至
-ꢀ
78℃后,向反应液中加入1.2个当量的甲醇钠(15.3mg,0.28mmol),升温至0℃下反应1 h。tlc监测至原料消失后,向反应液中加入饱和氯化铵溶液,乙酸乙酯萃取后经柱层析分离得到化合物19。1h nmr(400mhz,cdcl3)δ6.21(d,j=12.4hz,1h),5.87(d,j=13.3hz,1 h),4.78(m,1h),4.73(m,1h),3.89(m,1h),3.50(s,3h),2.66(dd,j=16.2,4.5hz,1h),2.31 (m,1h),1.64(s,3h),0.95(s,9h),0.89(s,6h),0.08(s,6h);
13
c nmr(100mhz,cdcl3)δ 176.2,143.7,126.2,122.0,112.5,76.0,55.6,52.3,45.6,33.7,26.8,23.5,19.6,18.7,-4.5,-4.8.
[0148]
实施例十八:化合物20的合成
[0149]
将乙基磷酸二乙酯(71.6μl,0.44mmol)溶解在四氢呋喃(10ml)中并冷却至-78℃, n2保护下加入3个当量的正丁基锂(0.46ml,1.1mmol)。将化合物17(120mg,0.37mmol) 溶解在四氢呋喃(5ml)溶液中,并降温至0℃。n2保护下,将乙基磷酸二乙酯的四氢呋喃溶液缓慢
滴入至实施例十七中的化合物19的四氢呋喃溶液中,保持反应液在该温度下反应1 h后,加入饱和氯化铵淬灭反应,柱层析分离后得到化合物20。1h nmr(400mhz,cdcl3)δ 6.23(d,j=12.5hz,1h),5.90(d,j=14.2hz,1h),4.92(m,1h),4.73(m,1h),4.12(m,4h), 3.82(t,j=3.5hz,1h),3.10(dd,j=13.0,3.5hz,1h),2.56(m,1h),1.60(s,3h),1.27(t,j=6.4 hz,6h),0.98(s,12h),0.82(s,3h),0.82(s,3h);
13
c nmr(100mhz,cdcl3)δ202.3,132.2, 126.2,121.0,112.3,77.8,65.5,65.2,55.3,54.1,46.3,41.1,38.6,26.7,24.5,19.8,19.6,16.3,13.8,
-ꢀ
4.7,-4.9.
[0150]
实施例十九:化合物21的合成
[0151]
将化合物11-a(34.5mg,0.07mmol)溶解在四氢呋喃(1ml)中,降温至0℃后,n2保护下向反应液中加入nah(6.18mg,60%in oil,0.15mmol)。10min后,向反应液中加入实施例十八中的化合物20(20mg,0.05mmol)的四氢呋喃(1ml)溶液。将反应液升温至50℃下回流反应12h后,加入饱和氯化铵溶液淬灭反应。反应液经乙酸乙酯萃取后经柱层析分离得到化合物21。1h nmr(400mhz,cdcl3)δ8.04(d,j=7.4hz,2h),7.66(t,j=7.2hz,1h), 7.43(m,2h),7.20(m,2h),5.90(m,1h),5.41(d,j=11.0hz,1h),4.87(m,1h),4.64(m,1h), 4.42(dd,j=8.7,3.4hz,1h),4.23(m,1h),3.72(ddd,j=11.8,7.0,5.4hz,1h),3.32(t,j=2.3 hz,1h),3.20(m,1h),3.11(m,1h),2.87(dd,j=10.2,5.4hz,1h),2.56(m,1h),2.22(m,1h), 1.68(s,3h),1.53(s,3h),0.99(s,18h),0.97(s,6h),0.86(d,j=6.5hz,3h),0.05(s,6h);
13
c nmr(100mhz,cdcl3)δ200.2,178.2,166.0,152.3,136.5,133.2,133.3,131.1,130.2,129.1,128.7, 123.7,121.9,112.2,80.2,82.1,79.2,65.2,55.1,45.5,44.5,40.2,38.1,22.1,18.5,16.2,11.9,12.0,
-ꢀ
4.9,-5.2,-5.7,-5.8.
[0152]
实施例二十:化合物22的合成
[0153]
将实施例十九中得到的化合物21(20mg,0.028mmol)溶解在甲苯(10ml)中加入至耐压管中,向反应液中加入催化量的grubbs ii催化剂(0.2mg)。将反应液加热回流过夜,tlc 监测至原料不再减少后,停止加热。将反应液减压浓缩后经柱层析分离得到化合物22。1h nmr(400mhz,cdcl3)δ8.12(d,j=7.4hz,2h),7.68(t,j=7.2hz,1h),7.45(m,2h),6.60 (m,1h),5.87(d,j=15.2hz,1h),5.41(m,2h),4.87(ddd,j=12.5,3.9,2.0,1h),3.64(m,1h), 3.45(dd,j=8.7,3.4hz,1h),3.24(m,1h),2.84(m,1h),2.56(m,2h),1.84(m,1h),1.78(d,j =2.0hz,3h),1.55(d,j=1.6hz,3h),0.98(s,9h),0.95(s,9h),0.94(s,6h),0.87(d,j=6.5 hz,1h),0.08(s,6h);
13
c nmr(100mhz,cdcl3)δ205.0,178.5,162.4,158.2,142.5,133.9, 132.2,130.3,129.3,128.3,127.3,122.64,86.2,84.6,79.2,65.4,56.4,52.1,43.5,40.2,35.6,25.6, 22.5,18.6,13.0,14.5,-4.8,-4.9,-5.1,-5.3.
[0154]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一类以ent-贝叶烷型二萜为手性臂的分子钳化合物及其制备方法和应用
- 含有ent-贝叶烷骨架的手性冠醚及其制备方法和应用
- 含有ent-贝叶烷骨架的开链手性冠醚及其制备和应用
- 制备分子印迹过氧化聚吡咯/纳米金修饰电极及其应用于电化学识别半胱氨酸对映体的制作方法
- 一类以(1s,2s)-1,2-环己二胺为隔离基、以异斯特维醇为手性臂的分子钳化合物及其制 ...的制作方法
- 一种l-脯氨酸类有机小分子催化剂及其制备和应用
- 一种d-乳酸脱色的新工艺的制作方法
- 钻孔用辅导片的感旋光性涂覆树脂及其应用
- 手性nd-322,nd-364或nd-364亚砜类似物及其制备方法
- 基于壳聚糖/海藻酸钠的手性传感器对含有锌离子的色氨酸对映体的手性识别的制作方法