一种水溶性炔酰胺缩合剂及其制备方法和应用与流程
[0001]
本发明涉及一种炔酰胺缩合剂及其应用,具体涉及一种其副产物能够通过水洗除去的水溶性炔酰胺缩合剂及其制备方法和应用,属于有机合成化学技术领域。
背景技术:
[0002]
酰胺键广泛存在于各种功能材料、医药、农药及各种精细化学品中,是有机化学中非常重要的一类官能团。酰胺键是蛋白质的基本结构单元,多肽和蛋白质是由天然氨基酸通过酰胺键按照某种特定的顺序连接起来的具有重要功能活性的生物大分子,对各种生命活动起着重要的调节作用。传统的形成酰胺键或肽键的方法主要是通过活化试剂或缩合剂将羧酸活化形成酰氯、酸酐、活化酯、酰基叠氮等活泼中间体,然后与胺发生亲核取代反应形成酰胺或者多肽。在各种形成肽键的方法中,缩合剂法是目前多肽合成中应用最为广泛的一种方法。
[0003]
n,n
’-
二环己基碳二亚胺(dcc)是由sheehan于1955年开发出来的第一个碳二亚胺型缩合剂(j.am.chem.soc.1955,77,1067-1068),至今仍是最常用的缩合剂之一,但是反应生成的副产物n,n
’-
二环己基脲(dcu)在大多数溶剂中的溶解性都很差,即便是通过离心、柱层析的方式也很难将其从目标产物中完全除去。为了使反应的后处理简单,人们开发出溶解性更好的碳二亚胺缩合剂,如n,n'-二异丙基碳二亚胺(dic)(j.chem.soc.,chem.commun.1981,543-545),以及副产物可通过水洗除去的碳二亚胺缩合剂,如1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edci)(j.org.chem.1961,26,2525-2528)、1,3-二(2,2-二甲基-1,3-二氧戊环-4-基甲基)碳二亚胺(bddc)(j.org.chem.1994,59,7503-7507)。然而,碳二亚胺型缩合剂在进行多肽缩合时,往往会导致较大程度的消旋,因此人们开发出消旋抑制剂与碳二亚胺型缩合剂配合使用,目前比较常用的有n-羟基丁二酰亚胺(hosu)(j.am.chem.soc.1964,86,1839-1842)、1-羟基苯并三唑(hobt)(chem.ber.1970,103,788-798)、n-羟基-7-氮杂苯并三氮(hoat)(j.am.chem.soc.1993,115,4397-4398)等。为了使用方便,众多科学家们将消旋抑制剂与缩合剂的功能设计到同一个分子上,如磷盐类缩合剂、脲盐/铵盐类缩合剂、亚胺盐类缩合剂等。其中1h-苯并三唑-1-基氧三吡咯烷基六氟磷酸盐(pybop)(tetrahedron lett.1990,31,205-208)、苯并三氮唑-n,n,n',n'-四甲基脲六氟磷酸盐(hbtu)(tetrahedron lett.1978,19,1269-1272)、n,n,n',n'-四甲基-o-(7-氮杂苯并三唑-1-基)六氟磷酸脲(hatu)(tetrahedron lett.1994,35,2279-2282)等缩合剂具有缩合效率高、消旋程度小等优点,在多肽合成领域得到了广泛应用。迄今为止已经有许多的缩合剂被开发出来,然而,绝大多数常用缩合剂都需要加入当量或者过量的碱和消旋抑制剂才能够实现较好的反应效果,这些额外的添加剂大大降低了多肽合成的原子经济性,不仅提高了多肽合成的成本,对环境保护也造成了很大的压力。
[0004]
2016年,赵军锋课题组设计开发出新型炔酰胺类缩合剂用于酰胺键和肽键的合成(j.am.chem.soc.2016,138,13135-13138)。这种新型缩合剂具有容易制备、稳定性好、分子量小、反应条件温和、使用过程中无需任何添加剂等优点,更重要的是α-手性酸在缩合过程
中不发生消旋,大大提高了产物的纯度和收率。但现有的炔酰胺类缩合剂促进的酰胺化反应必须通过柱层析的方法来分离和纯化产物,造成成本较高。
技术实现要素:
[0005]
本发明在现有技术的基础上对炔酰胺的结构进行了改进,开发了其副产物可通过水洗除去的炔酰胺缩合剂。新型的水溶性炔酰胺缩合剂除了具有容易制备、无需额外的添加剂、不诱发消旋、底物适用范围广等优点,反应完全后还能通过弱酸水解即可使副产物溶于水中,水洗即可将副产物除去,可简化产物后处理工序。
[0006]
根据本发明的第一种实施方案,提供一种水溶性炔酰胺缩合剂。
[0007]
一种水溶性炔酰胺缩合剂,其特征在于:该炔酰胺缩合剂具有如下式(i)的结构:
[0008][0009]
其中,r选自甲基磺酰基、苯磺酰基、对甲苯磺酰基、三氟乙酰基及其他吸电子基团。
[0010]
根据本发明的第二种实施方案,提供一种制备具有式(i)结构的水溶性炔酰胺缩合剂的方法,该方法包括以下步骤:
[0011]
1)将具有式(ii)结构的n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)酰胺与二氯乙烯在溶剂i中混合,
[0012]
2)向步骤1)的混合物中加入碱,进行反应,分离,获得具有式(i)结构的水溶性炔酰胺缩合剂;具体反应为:
[0013][0014]
其中,r选自甲基磺酰基和对甲苯磺酰基。
[0015]
优选的是,步骤1)中所述溶剂i为有机溶剂,优选所述溶剂i为二甲亚砜、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮中的一种或多种。
[0016]
优选的是,具有式(ii)结构的n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)酰胺与二氯乙烯加入的摩尔比为1:0.8-5,优选为1:1-3;更优选为1:1.1-2。
[0017]
优选的是,步骤2)中所碱为碱为nah、cah2、t-buona、koh、naoh、etona、etoli、cs2co3、k2co3、na2co3、ca(oh)2、lioh、dbu中的一种或多种。
[0018]
优选的是,步骤2)反应温度为15-100℃,优选为20-90℃,更优选为25-80℃。反应时间为0.2-48h,优选为0.5-36h,更优选为1-24h。
[0019]
优选的是,步骤2)所述分离采用过滤、离心或柱层析。
[0020]
优选的是,步骤2)碱的加入量与具有式(ii)结构的n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)酰胺加入量的摩尔比为1-10:1,优选为2-8:1;更优选为3-6:1。
[0021]
根据本发明的第三种实施方案,提供一种水溶性炔酰胺缩合剂的应用。
[0022]
一种水溶性炔酰胺缩合剂的应用,其特征在于:将具有式(i)结构的水溶性炔酰胺缩合剂用于合成酰胺、多肽、脂类化合物或硫酯类化合物。
[0023]
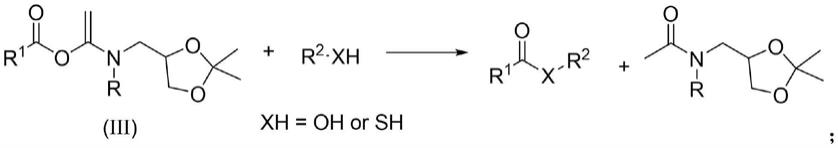
优选的是,所述将所述水溶性炔酰胺缩合剂用于合成酯类化合物或硫酯类化合物的方法具体为:
[0024]
1a)将羧酸类化合物与具有结构通式(i)的水溶性炔酰胺缩合剂在溶剂ii中进行反应,获得具有式(iii)结构的化合物;具体反应为:
[0025][0026]
2a)步骤1)反应完成后,将获得的具有式(iii)结构的化合物溶于溶剂iii中,加入醇类、酚类、硫醇类、硫酚类化合物中的一种,并加入催化剂,搅拌反应,获得含有酯类化合物或硫酯类化合物的混合物;具体反应为:
[0027][0028]
3a)向步骤2)获得的含有酯类化合物或硫酯类化合物的混合物中加入稀酸水溶液,水解未反应的缩合剂和反应得到的副产物,分离,获得目标酯类化合物或硫酯类化合物;其反应式为:
[0029][0030]
式中,r1选自烷基、环烷基、烯基、炔基、芳基、杂环基、杂环芳基、保护的α-氨基烷基、保护的β-氨基烷基、保护的多肽链烷基中的一种,r选自甲基磺酰基、苯磺酰基、对甲苯磺酰基、三氟乙酰基及其他吸电子基团中的一种,r2选自脂肪族取代基团、芳香族取代基团。
[0031]
优选的是,步骤1a)中,所述羧酸类化合物为由烃基和羧基相连构成的有机酸。
[0032]
优选的是,步骤1a)所述羧酸类化合物优选为脂肪酸、芳香酸、杂环酸、炔酸、烯酸、α-氨基酸、β-氨基酸中的一种或多种,所述羧酸类化合物更优选为甲酸、乙酸、苯乙酸、石胆酸及其他脂肪族羧酸、丙炔酸、苯丙炔酸、肉桂酸、丙烯酸及其他不饱和酸、苯甲酸、对甲基苯甲酸、对氯苯甲酸、吡啶-2-甲酸、呋喃-2-甲酸及其他芳香酸、苄氧羰基保护氨基的α-氨基酸、叔丁氧羰基保护氨基的α-氨基酸、芴甲氧羰基保护氨基的α-氨基酸、乙酰基保护氨基的α-氨基酸以及多肽羧酸。
[0033]
优选的是,步骤1a)羧酸类化合物与具有结构通式(i)的水溶性炔酰胺缩合剂的加
入量的摩尔比为1:1-5,优选为1:1.1-4,更优选为1:1.2-3。
[0034]
优选的是,步骤1a)所述溶剂ii为二氯甲烷、水、氯仿、1,2-二氯乙烷中的任一种,或所述溶剂ii为水与二甲亚砜的混合物、水与n,n-二甲基甲酰胺的混合物。
[0035]
优选的是,步骤2a)中,所述醇类、酚类、硫醇类、硫酚类化合物为官能团为-oh或-sh的有机化合物。
[0036]
优选的是,步骤2a)所述醇类、酚类、硫醇类、硫酚类化合物优选为脂肪醇、芳香醇中官能团为-oh、-sh的有机化合物,所述醇类、酚类、硫醇类、硫酚类化合物更优选为乙醇、三氟乙醇、丙醇、丁醇、异丙醇及其他脂肪族伯醇和仲醇、苯酚、雌酚酮、对甲氧基苯酚、对氯苯酚、对硝基苯酚及其他取代的苯酚、乙硫醇、正己硫醇、环己硫醇、叔丁硫醇等脂肪族硫醇、4-巯基苯甲酸、对甲基苯硫酚、对氯苯硫酚、对溴苯硫酚及其他取代的硫酚、氨基保护的半胱氨酸酯、多肽侧链的巯基中的任一种。
[0037]
优选的是,步骤2a)所述醇类、酚类、硫醇类、硫酚类化合物与羧酸类化合物的加入量的摩尔比为1:1-20,优选为1:1.5-15,更优选为1:2-10。
[0038]
优选的是,步骤2a)所述溶剂iii为水、乙腈、二甲亚砜、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮、乙腈与水的混合物、水与二甲亚砜的混合物、水与n,n-二甲基甲酰胺的混合物中的一种或多种。
[0039]
优选的是,步骤2a)所述催化剂为三乙胺或n,n-二异丙基乙胺。
[0040]
优选的是,步骤2a)所述催化剂与羧酸类化合物的加入量的摩尔比为0.01-10:1,优选为0.02-5:1,更优选为0.03-1:1。
[0041]
优选的是,步骤3a)中,所述稀酸为稀硫酸、稀盐酸、磷酸、醋酸、柠檬酸中的一种。所述稀酸的浓度为0.01~5mol/l,优选为0.5~2mol/l,更优选为0.1~1mol/l。
[0042]
优选的是,步骤1a)具体为:在反应器中加入羧酸类化合物、具有结构通式(i)的水溶性炔酰胺缩合剂、溶剂ii,混合,在温度为0-60℃(优选为5-50℃)、搅拌的条件下进行反应,反应结束后,除去溶剂ii,获得具有式(iii)结构的化合物。
[0043]
优选的是,步骤2a)具体为:步骤1)反应完成后,将步骤1a)获得的具有式(iii)结构的化合物溶入溶剂iii中,加入醇类、酚类、硫醇类、硫酚类化合物中的一种,然后加入催化剂,搅拌,再温度为0-60℃(优选为5-50℃)、搅拌的条件下进行反应,获得含有酯类化合物或硫酯类化合物与副产物的混合物。
[0044]
优选的是,步骤3a)具体为:向步骤2)获得的含有酯类化合物或硫酯类化合物的混合物中任选地加入稀释剂进行稀释(作为优选,所述稀释剂为二氯甲烷或乙酸乙酯),然后加入稀酸进行洗涤,水解未反应的缩合剂和反应得到的副产物,产物析出,过滤水洗,获得目标酯类化合物或硫酯类化合物。
[0045]
优选的是,所述将所述水溶性炔酰胺缩合剂用于合成酰胺化合物或多肽类化合物的方法,具体为:
[0046]
1b)将羧酸类化合物与具有结构通式(i)的水溶性炔酰胺缩合剂在溶剂ii中进行反应,获得具有式(iv)结构的化合物;具体反应为:
[0047][0048]
2b)步骤1b)反应完成后,向获得的具有式(iv)结构的化合物中加入胺类化合物,搅拌反应,获得含有酰胺类化合物或多肽类化合物与副产物的混合物;具体反应为:
[0049][0050]
3b)向步骤2b)获得的含有酰胺类化合物或多肽类化合物与副产物的混合物中加入稀释液,稀酸水溶液,水解未反应的缩合剂和反应得到的副产物,分离,获得目标酰胺类化合物或多肽类化合物;其反应式为:
[0051][0052]
式中,r1选自烷基、环烷基、烯基、炔基、芳基、杂环基、杂环芳基、保护的α-氨基烷基、保护的β-氨基烷基、保护的多肽链烷基中的一种,r选自甲基磺酰基、苯磺酰基、对甲苯磺酰基、三氟乙酰基及其他吸电子基团中的一种,r3、r4选自脂肪族取代基团、芳香族取代基团。
[0053]
优选的是,步骤1b)中,所述羧酸类化合物为由烃基和羧基相连构成的有机酸。
[0054]
优选的是,步骤1b)所述羧酸类化合物优选为脂肪酸、芳香酸、杂环酸、炔酸、烯酸、α-氨基酸、β-氨基酸中的任一种,所述羧酸类化合物更优选为甲酸、乙酸、苯乙酸、石胆酸及其他脂肪族羧酸、丙炔酸、苯丙炔酸、肉桂酸、丙烯酸及其他不饱和酸、苯甲酸、对甲基苯甲酸、对氯苯甲酸、吡啶-2-甲酸、呋喃-2-甲酸及其他芳香酸、苄氧羰基保护氨基的α-氨基酸、叔丁氧羰基保护氨基的α-氨基酸、芴甲氧羰基保护氨基的α-氨基酸、乙酰基保护氨基的α-氨基酸以及多肽羧酸。
[0055]
优选的是,步骤1b)羧酸类化合物与具有结构通式(i)的水溶性炔酰胺缩合剂的加入量的摩尔比为1:1-5,优选为1:1.1-4,更优选为1:1.2-3。
[0056]
优选的是,步骤1b)所述溶剂ii为有机溶剂。
[0057]
优选的是,步骤1b)所述溶剂ii优选为二氯甲烷、水、氯仿、1,2-二氯乙烷中的任一种,或所述溶剂ii为水与二甲亚砜的混合物、水与n,n-二甲基甲酰胺的混合物。
[0058]
优选的是,步骤2b)中,所述胺类化合物为一级胺或二级胺,所述胺类化合物优选为脂肪族伯胺、脂肪族仲胺、芳胺、α-氨基酸甲酯、α-氨基酸乙酯、α-氨基酸叔丁酯、α-氨基酸苄酯、β-氨基酸甲酯、β-氨基酸乙酯、β-氨基酸叔丁酯、β-氨基酸苄酯中的任一种。
[0059]
优选的是,步骤2b)所述羧酸类化合物与胺类化合物的加入量的摩尔比为1:1-5,优选为1:1.1-4,更优选为1:1.2-3。
[0060]
优选的是,步骤3b)中,所述稀酸为稀硫酸、稀盐酸、磷酸、醋酸、柠檬酸中的任一
种。
[0061]
优选的是,步骤3b)所述稀酸的浓度为0.01~5mol/l,优选为0.5~2mol/l,更优选为0.1~1mol/l。
[0062]
优选的是,步骤1b)具体为:在反应器中加入羧酸类化合物、具有结构通式(i)的水溶性炔酰胺缩合剂、溶剂ii,混合,在温度为0-60℃(优选为5-50℃)、搅拌的条件下进行反应,获得具有式(iv)结构的化合物。
[0063]
优选的是,步骤2b)具体为:步骤1b)反应完成后,向步骤1b)获得的具有式(iv)结构的化合物中加入胺类化合物,在温度为0-60℃(优选为5-50℃)、搅拌的条件下进行反应,获得含有酰胺类化合物或多肽类化合物与副产物的混合物。
[0064]
优选的是,步骤3b)具体为:向步骤2b)获得的含有酰胺类化合物或多肽类化合物的混合物中加入稀释剂(作为优选,所述稀释剂为二氯甲烷或乙酸乙酯)。然后加入稀酸进行洗涤,水解未反应的缩合剂和反应得到的副产物,产物析出,过滤水洗,获得目标酰胺类化合物或多肽类化合物。
[0065]
优选的是,步骤3b)具体为:除去步骤2b)获得的含有酰胺类化合物或多肽类化合物的混合物中的溶剂,加入溶剂ⅳ(作为优选,所述溶剂ⅳ为甲醇、乙醇、异丙醇或乙腈中的一种)。然后加入稀酸进行洗涤,水解未反应的缩合剂和反应得到的副产物,产物析出,过滤水洗,获得目标酰胺类化合物或多肽类化合物。
[0066]
在本发明中,具有式(ii)结构的n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)酰胺具体为:n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)甲磺酰胺(结构式如制备实施例1)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)苯磺酰胺(结构式如制备实施例2)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)对甲苯磺酰胺(结构式如制备实施例3)或n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)三氟乙酰胺(结构式如制备实施例4)。
[0067]
在本发明中,n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)甲磺酰胺(结构式如制备实施例1)通过以下方法制备获得:
[0068]
在冰浴条件下,将(2,2-二甲基-[1,3]-二氧杂环戊烷-4-基)-甲胺与3倍量的三乙胺混合在二氯甲烷溶液中进行搅拌,缓慢滴加与(2,2-二甲基-[1,3]-二氧杂环戊烷-4-基)-甲胺当量的甲基磺酰氯,tcl检测反应,反应完全后加水,水相使用二氯甲烷萃取,合并有机相,饱和食盐水洗涤,分出有机相,用无水硫酸钠干燥,浓缩有机相即得到n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)甲磺酰胺。
[0069]
在本发明中,n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)苯磺酰胺(结构式如制备实施例2)通过以下方法制备获得:
[0070]
在冰浴条件下,将(2,2-二甲基-[1,3]-二氧杂环戊烷-4-基)-甲胺与3倍量的三乙胺混合在二氯甲烷溶液中进行搅拌,缓慢滴加与(2,2-二甲基-[1,3]-二氧杂环戊烷-4-基)-甲胺当量的苯磺酰氯,tcl检测反应,反应完全后加水,水相使用二氯甲烷萃取,合并有机相,饱和食盐水洗涤,分出有机相,用无水硫酸钠干燥,浓缩有机相即得到n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)苯磺酰胺。
[0071]
在本发明中,n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)对甲苯磺酰胺(结构式如制备实施例3)通过以下方法制备获得:
[0072]
在冰浴条件下,将(2,2-二甲基-[1,3]-二氧杂环戊烷-4-基)-甲胺与3倍量的三乙
胺混合在二氯甲烷溶液中进行搅拌,缓慢滴加与(2,2-二甲基-[1,3]-二氧杂环戊烷-4-基)-甲胺当量的对甲苯磺酰氯,tcl检测反应,反应完全后加水,水相使用二氯甲烷萃取,合并有机相,饱和食盐水洗涤,分出有机相,用无水硫酸钠干燥,浓缩有机相即得到n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)对甲苯酰胺。
[0073]
在本发明中,n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)三氟乙酰胺(结构式如制备实施例4)通过以下方法制备获得:
[0074]
在冰浴条件下,将(2,2-二甲基-[1,3]-二氧杂环戊烷-4-基)-甲胺与3倍量的三乙胺混合在二氯甲烷溶液中进行搅拌,缓慢滴加与(2,2-二甲基-[1,3]-二氧杂环戊烷-4-基)-甲胺当量的三氟乙酰氯,tcl检测反应,反应完全后加水,水相使用二氯甲烷萃取,合并有机相,饱和食盐水洗涤,分出有机相,用无水硫酸钠干燥,浓缩有机相即得到n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)三氟乙酰胺。
[0075]
在本发明中,第一步生成α-酰氧基烯酰胺类化合物的反应在n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、1,4-二氧六环和四氢呋喃等溶剂中,基本上没有反应。在丙酮、乙腈、甲醇中,反应可以发生,但是比较慢。在卤代烃溶剂中反应效果最好,如二氯甲烷、1,2-二氯甲烷和氯仿等溶剂中,反应在室温条件下1-2个小时即可完成,产物收率几乎可以达到100%。
[0076]
在本发明中,α-酰氧基烯酰胺类化合物是一种活化的酯,其可与一级或者二级胺顺利地在室温条件下发生反应来制备酰胺和多肽。羧酸与炔酰胺的加成反应与α-酰氧基烯酰胺类化合物的酰胺化反应可以分步进行,也可以通过“一锅法”来实现。
[0077]
在本发明中,对炔酰胺缩合剂引入了缩酮的结构(2,2-二甲基-1,3-二氧戊环-4-基),该结构在弱酸性的条件下即可发生水解,生成两个羟基,使炔酰胺缩合剂及其副产物的水溶性极大地增加。因此,在反应完全后生成的副产物和多余炔酰胺缩合剂经弱酸水解,可使副产物和多余炔酰胺缩合剂溶于水中,并通过水洗将副产物除去,实现产物的简便分离,可避免进行柱层析色谱分离纯化等操作。使用本发明的技术方案,制备获得的水溶性炔酰胺缩合剂用于制备酰胺、多肽、酯及硫酯等化合物,其纯度可以达到95~99%。其中,通过该方案制备的具有手性的酰胺、多肽、酯及硫酯类化合物的光学纯度可达到99%以上。
[0078]
另外,该缩合剂在使用过程中无需额外的添加剂及碱,在温和的条件下,以“一锅法”即可实现酰胺和多肽化合物、酯和硫酯类化合物的合成。尤其是在天然α-氨基酸以及其他手性酸的反应中,保持产物的手性纯度是非常重要的,该水溶性炔酰胺缩合剂能够有效地控制合成过程中手性酸消旋的问题。因此,反应之后仅需通过弱酸水洗就能够使实现产物与副产物的分离,无需柱层析色谱分离纯化等操作,即可得到纯度相当高的产物,使酰胺和多肽化合物、酯和硫酯类化合物的合成更加简洁、方便,而且对环境影响小,非常绿色环保。
[0079]
与现有技术相比,本发明具有以下有益效果:
[0080]
1、相对于传统的缩合剂dcc、pybop、hbtu,本发明所述的新型水溶性炔酰胺缩合剂具有较好的性能,能够高效方便地制备酰胺、多肽、酯和硫酯类化合物。
[0081]
2、反应完全后还能通过弱酸水解即可使副产物溶于水中,水洗即可将副产物除去,可简化产物后处理工序。
[0082]
3、本发明所述的新型水溶性炔酰胺缩合剂在促成酰胺键与酯键形成过程中无需额外的添加剂,提升了反应的原子经济性。
[0083]
4、本发明所述的用新型水溶性炔酰胺缩合剂制备酰胺、多肽、酯和硫酯类化合物,在后处理过程中使用水纯化产物,避免了过多有机溶剂的使用,对环境影响小,非常绿色环保。
具体实施方式
[0084]
下面对本发明的技术方案进行举例说明,本发明请求保护的范围包括但不限于以下实施例。
[0085]
制备实施例1
[0086]
n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)甲磺酰胺的制备:
[0087]
在冰浴条件下,将5mmol(2,2-二甲基-[1,3]-二氧杂环戊烷-4-基)-甲胺与15mmol的三乙胺混合在二氯甲烷溶液中进行搅拌,缓慢滴加5mol甲基磺酰氯,tcl检测反应,反应完全后加水,水相使用二氯甲烷萃取两次,合并有机相,饱和食盐水洗涤一次,分出有机相,用无水硫酸钠干燥,浓缩有机相即得到n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)甲磺酰胺,淡黄色液体,产率98%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0088][0089]1h nmr(400mhz,cdcl3)δ4.73(s,1h),4.28(qd,j=6.3,3.9hz,1h),4.07(dd,j=8.5,6.5hz,1h),3.75(dd,j=8.5,6.0hz,1h),3.34(ddd,j=13.3,6.7,3.8hz,1h),3.23
–
3.16(m,1h),3.00(s,3h),1.44(s,3h),1.35(s,3h).
[0090]
13
c nmr(100mhz,cdcl3)δ=109.9,74.5,66.6,45.6,40.7,26.9,25.3.
[0091]
hrms(esi)m/z calcd.for c7h
16
no4s[m+h]
+
:210.0795,found:210.0800。
[0092]
制备实施例2
[0093]
n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)苯磺酰胺的制备:
[0094]
在冰浴条件下,将5mmol(2,2-二甲基-[1,3]-二氧杂环戊烷-4-基)-甲胺与15mmol的三乙胺混合在二氯甲烷溶液中进行搅拌,缓慢滴加5mol苯磺酰氯,tcl检测反应,反应完全后加水,水相使用二氯甲烷萃取两次,合并有机相,饱和食盐水洗涤一次,分出有机相,用无水硫酸钠干燥,浓缩有机相即得到n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)苯磺酰胺,白色固体,产率95%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0095][0096]1h nmr(400mhz,cdcl3)δ7.90
–
7.83(m,2h),7.80(s,1h),7.66
–
7.56(m,3h),4.05(p,j=7.0hz,1h),3.96(dd,j=11.4,7.0hz,1h),3.71(dd,j=11.4,6.9hz,1h),3.59(dd,j=12.4,7.0hz,1h),3.39(dd,j=12.4,7.0hz,1h),1.39(s,3h),1.34(s,3h).
[0097]
13
c nmr(100mhz,cdcl3)δ140.0,133.8,129.5,128.2,109.4,74.6,67.0,46.7,25.5.
[0098]
hrms(esi)m/z calcd.for c
12
h
18
no4s[m+h]
+
:272.0951,found:272.0959。
[0099]
制备实施例3
[0100]
在本发明中,n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)对甲苯磺酰胺的制备:
[0101]
在冰浴条件下,将5mmol(2,2-二甲基-[1,3]-二氧杂环戊烷-4-基)-甲胺与15mmol的三乙胺混合在二氯甲烷溶液中进行搅拌,缓慢滴加5mol对甲苯磺酰氯,tcl检测反应,反应完全后加水,水相使用二氯甲烷萃取两次,合并有机相,饱和食盐水洗涤一次,分出有机相,用无水硫酸钠干燥,浓缩有机相即得到n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)对甲苯磺酰胺,白色固体,产率97%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0102][0103]1h nmr(400mhz,cdcl3)δ7.75(d,j=8.3hz,2h),7.31(d,j=8.1hz,2h),4.94
–
4.77(m,1h),4.22
–
4.14(m,1h),3.99(dd,j=8.4,6.4hz,1h),3.68(dd,j=8.5,6.0hz,1h),3.13(dddd,j=12.8,7.0,4.1,1.6hz,1h),3.00
–
2.93(m,1h),2.43(s,3h),1.35(s,3h),1.30(s,3h).
[0104]
13
c nmr(100mhz,cdcl3)δ143.7,136.9,129.9,127.2,109.8,74.1,66.7,45.4,26.9,25.3,21.6.
[0105]
hrms(esi)m/z calcd.for c
13
h
20
no4s[m+h]
+
:286.1108,found:286.1113。
[0106]
制备实施例4
[0107]
在本发明中,n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)三氟乙酰胺的制备:
[0108]
在冰浴条件下,将5mmol(2,2-二甲基-[1,3]-二氧杂环戊烷-4-基)-甲胺与15mmol的三乙胺混合在二氯甲烷溶液中进行搅拌,缓慢滴加5mol三氟乙酰氯,tcl检测反应,反应完全后加水,水相使用二氯甲烷萃取两次,合并有机相,饱和食盐水洗涤一次,分出有机相,用无水硫酸钠干燥,浓缩有机相即得到n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)三氟乙酰胺,淡黄色液体,产率92%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0109][0110]1h nmr(400mhz,cdcl3)δ6.69(s,1h),4.52(p,j=7.0hz,1h),3.96(dd,j=11.4,7.0hz,1h),3.80(dd,j=12.4,7.1hz,1h),3.71(dd,j=11.5,7.0hz,1h),3.46(dd,j=12.4,7.0hz,1h),1.39(s,3h),1.34(s,3h).
[0111]
13
c nmr(100mhz,cdcl3)δ159.2,159.0,158.7,158.5,118.7,116.5,114.4,112.2,109.4,74.6,67.0,43.1,43.1,43.1,25.5.
[0112]
hrms(esi)m/z calcd.for c8h
13
f3no3[m+h]
+
:228.0842,found:228.088.
[0113]
制备实施例5
[0114]
n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基甲磺酰胺的制备:
[0115]
将n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)甲磺酰胺与3倍量的1,1-二氯乙烯在有机溶剂中混合,加入以相当于n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)甲磺酰胺的摩尔量5倍量的碱,反应温度70℃,tlc点板检测,反应结束后,反应液加冰水后用乙酸乙酯萃取3次,有机层浓缩后经柱层析分离得到纯的n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙
炔基甲磺酰胺,白色固体,产率95%。以下是产物的结构式及核磁表征数据、质谱表征数据:
[0116][0117]1h nmr(400mhz,dmso)δ4.33(ddd,j=12.2,6.8,5.0hz,1h),4.05(dd,j=8.7,6.4hz,1h),3.86(s,1h),3.73(dd,j=8.7,5.1hz,1h),3.49(qd,j=14.1,6.1hz,2h),3.25(s,3h),1.36(s,3h),1.28(s,3h).
[0118]
13
c nmr(100mhz,dmso)δ109.1,75.9,72.7,66.0,61.3,53.5,37.9,26.6,25.2.
[0119]
hrms(esi)m/z calcd.for c9h
16
no4s[m+h]
+
:234.0795,found:234.0797。
[0120]
制备实施例6
[0121]
n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基苯磺酰胺的制备:
[0122]
将n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)苯磺酰胺与3倍量的1,1-二氯乙烯在有机溶剂中混合,加入以相当于n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)苯磺酰胺的摩尔量5倍量的碱,反应温度70℃,tlc点板检测,反应结束后,反应液加冰水后用乙酸乙酯萃取3次,有机层浓缩后经柱层析分离得到纯的n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基苯磺酰胺,白色固体,产率94%。以下是产物的结构式及核磁表征数据、质谱表征数据:
[0123][0124]1hnmr(500mhz,chloroform-d)δ7.86(d,j=7.6hz,2h),7.62(t,j=7.5hz,1h),7.52(t,j=7.4hz,2h),4.41
–
4.30(m,1h),4.10(dd,j=8.8,6.1hz,1h),3.89(dd,j=8.8,5.2hz,1h),3.53(dd,j=13.4,5.4hz,1h),3.30(dd,j=13.4,7.2hz,1h),2.73(s,1h),2.44(s,3h),1.40(s,3h),1.32(s,3h).
[0125]
13
c nmr(100mhz,cdcl3)δ138.1,133.8,129.1,127.9,109.9,76.4,73.5,67.4,59.2,53.7,27.0,25.4.
[0126]
hrms(esi)m/z calcd.for c
14
h
17
nnao4s[m+h]
+
:318.0770,found:318.0778。
[0127]
制备实施例7
[0128]
n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基对甲苯磺酰胺的制备:
[0129]
将n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)对甲苯磺酰胺与3倍量的1,1-二氯乙烯在有机溶剂中混合,加入以相当于n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)对甲苯磺酰胺的摩尔量5倍量的碱,反应温度70℃,tlc点板检测,反应结束后,反应液加冰水后用乙酸乙酯萃取3次,有机层浓缩后经柱层析分离得到纯的n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基对甲苯磺酰胺,白色固体,产率93%。以下是产物的结构式及核磁表征数据、质谱表征数据:
[0130]
[0131]1hnmr(400mhz,cdcl3)δ7.81(d,j=8.3hz,2h),7.36(d,j=8.0hz,2h),4.40
–
4.31(m,1h),4.09(dd,j=8.8,6.1hz,1h),3.87(dd,j=8.8,5.2hz,1h),3.51(dd,j=13.4,5.4hz,1h),3.32(dd,j=13.4,7.2hz,1h),2.75(s,1h),2.46(s,3h),1.41(s,3h),1.33(s,3h).
[0132]
13
c nmr(100mhz,cdcl3)δ145.1,134.4,130.0,127.9,109.9,76.4,73.5,67.4,59.2,53.7,27.0,25.4,21.8.
[0133]
hrms(esi)m/z calcd.for c
15
h
20
no4s[m+h]
+
:310.1108,found:310.1116。
[0134]
制备实施例8
[0135]
n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基三氟乙酰胺的制备:
[0136]
将n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)三氟乙酰胺与3倍量的1,1-二氯乙烯在有机溶剂中混合,加入以相当于n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)三氟乙酰胺的摩尔量5倍量的碱,反应温度70℃,tlc点板检测,反应结束后,反应液加冰水后用乙酸乙酯萃取3次,有机层浓缩后经柱层析分离得到纯的n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基三氟乙酰胺,淡黄色液体,产率90%。以下是产物的结构式及核磁表征数据、质谱表征数据:
[0137][0138]1hnmr(400mhz,cdcl3)δ4.52(p,j=7.0hz,1h),3.96(dd,j=11.4,7.0hz,1h),3.71(dd,j=11.5,7.0hz,1h),3.44(dd,j=12.4,7.0hz,1h),3.19(dd,j=12.4,7.0hz,1h),2.45(s,1h),1.39(s,3h),1.34(s,3h).
[0139]
13
c nmr(100mhz,cdcl3)δ160.98,160.72,160.47,160.21,117.71,115.57,113.43,111.29,109.38,77.87,77.84,77.81,77.78,74.09,66.98,57.51,45.46,45.45,45.43,25.55.
[0140]
hrms(esi)m/z calcd.for c
10
h
12
f3nnao3[m+h]
+
:274.0661,found:274.0657。
[0141]
应用实施例1
[0142]
取苯丙炔酸(0.5mmol)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基甲磺酰胺(0.55mmol)溶于3ml二氯甲烷中,在室温下搅拌反应,tlc监测反应过程,待苯丙炔酸消耗完全,加入2-羟基乙胺(0.55mmol),tlc监测反应过程,反应结束后加入15ml乙酸乙酯稀释反应液,用0.5m的稀盐酸15ml震摇洗涤有机相2次,分去水相,用无水硫酸镁干燥有机相,减压蒸馏除去溶剂,得到纯的产物,白色固体,产率90%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0143][0144]1h nmr(400mhz,cdcl3)δ7.51(d,j=7.0hz,2h),7.39(d,j=7.3hz,1h),7.33(t,j=7.4hz,2h),6.83(t,j=5.9hz,1h),3.80
–
3.75(m,2h),3.52(q,j=5.4hz,2h).
[0145]
13
c nmr(100mhz,cdcl3)δ154.5,132.6,130.3,128.6,120.2,85.6,82.9,61.5,
42.7.
[0146]
hrms(esi)m/z calcd.for c
12
h
12
o2[m+h]
+
:190.0868,found:190.0867。
[0147]
应用实施例2
[0148]
取苯丙炔酸(0.5mmol)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基甲磺酰胺(0.55mmol)溶于3ml二氯甲烷中,在室温下搅拌反应,tlc监测反应过程,待原料酸消耗完全,加入哌啶(0.55mmol),tlc监测反应过程,反应结束后加入15ml乙酸乙酯稀释反应液,用0.5m的稀盐酸15ml震摇洗涤有机相2次,分去水相,用无水硫酸镁干燥有机相,减压蒸馏除去溶剂,得到纯的产物,白色固体,产率96%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0149][0150]1h nmr(400mhz,cdcl3)δ7.54(d,j=8.1hz,2h),7.43
–
7.33(m,3h),3.80
–
3.75(m,2h),3.65
–
3.59(m,2h),1.71
–
1.64(m,4h),1.62
–
1.56(m,2h).
[0151]
13
c nmr(100mhz,cdcl3)δ153.05,132.41,129.95,128.57,120.85,90.37,81.59,48.33,42.49,26.56,25.50,24.65.
[0152]
hrms(esi)m/z calcd.for c
14
h
16
no[m+h]
+
:214.1226,found:214.1230。
[0153]
应用实施例3
[0154]
取甲基苯甲酸(0.5mmol)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基甲磺酰胺(0.55mmol)溶于3ml二氯甲烷中,在室温下搅拌反应,tlc监测反应过程,待原料酸消耗完全,加入四氢吡咯(0.55mmol),tlc监测反应过程,反应结束后减压蒸馏除去溶剂,加入少量的乙醇,加入0.5m的稀盐酸10ml,搅拌10min,产物析出,过滤水洗,收集固体,得到纯的产物,白色固体,产率96%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0155][0156]1h nmr(400mhz,cdcl3)δ7.42(d,j=8.0hz,2h),7.19(d,j=7.8hz,2h),3.64(t,j=6.7hz,2h),3.44(t,j=6.3hz,2h),2.37(s,3h),1.94(q,j=6.2hz,2h),1.87(q,j=6.1hz,2h).
[0157]
13
c nmr(100mhz,cdcl3)δ169.95,139.98,134.40,128.90,127.30,49.75,46.30,26.51,24.55,21.46.
[0158]
hrms(esi)m/z calcd.for c
12
h
16
no[m+h]
+
:190.1226,found:190.1225。
[0159]
应用实施例4
[0160]
取fmoc-ala-oh(0.5mmol)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基甲磺酰胺(0.55mmol)溶于5ml二氯甲烷中,在室温下搅拌反应,tlc监测反应过程,待原料酸消耗完全,加入h-phe-otbu(0.55mmol),tlc监测反应过程,反应结束后加入15ml乙酸乙酯稀释反应液,用0.2m的稀盐酸15ml震摇洗涤有机相3次,分去水相,用无水硫酸镁干燥有机相,浓缩后用二氯甲烷/石油醚重结晶,得到纯的产物,白色固体,产率87%。以下是产物的
结构式及核磁共振实验数据、质谱实验数据:
[0161][0162]1h nmr(400mhz,cdcl3)δ7.75(d,j=7.5hz,2h),7.58(d,j=7.0hz,2h),7.38(t,j=7.4hz,2h),7.29(t,j=7.4hz,2h),7.24
–
7.10(m,5h),6.69
–
6.48(m,1h),5.60
–
5.40(m,1h),4.73(q,j=6.1hz,1h),4.39(dd,j=10.3,7.3hz,1h),4.36
–
4.22(m,2h),4.19(t,j=7.0hz,1h),3.08(h,j=8.0,7.2hz,2h),1.38(s,9h),1.35(s,3h).
[0163]
13
c nmr(100mhz,cdcl3)δ171.84,170.35,155.94,143.92,141.37,136.09,129.56,128.44,127.80,127.16,127.05,125.17,120.06,82.51,67.20,53.74,50.51,47.21,38.07,28.02,18.87.
[0164]
hrms(esi)m/z calcd.for c
31
h
35
n2o5[m+h]
+
:515.2540,found:515.2547.dr>99:1。
[0165]
应用实施例5
[0166]
取fmoc-aib-oh(0.5mmol)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基甲磺酰胺(0.55mmol)溶于5ml的二氯甲烷中,在室温下搅拌反应,tlc监测反应过程,待原料酸消耗完全,加入h-ala-otbu(0.55mmol),tlc监测反应过程,反应结束后减压蒸馏除去溶剂,加入少量的乙醇,加入0.2m的稀盐酸20ml,搅拌10min,产物析出,过滤水洗,收集固体,得到纯的产物,白色固体,产率85%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0167][0168]1h nmr(400mhz,cdcl3)δ7.75(d,j=7.5hz,2h),7.61
–
7.57(m,2h),7.39(t,j=7.4hz,2h),7.31(t,j=7.4hz,2h),6.74(s,1h),5.52(s,1h),4.45
–
4.35(m,3h),4.20(t,j=6.7hz,1h),1.53(s,6h),1.44(s,9h),1.35(d,j=6.9hz,3h).
[0169]
13
c nmr(100mhz,cdcl3)δ173.82,172.23,155.11,144.00,143.96,141.42,127.78,127.17,125.15,125.12,120.06,82.13,66.72,56.84,49.02,47.33,28.06,25.71,25.27,18.54.
[0170]
hrms(esi)m/z calcd.for c
26
h
33
n2o5[m+h]
+
:453,2384,found:453.2380。
[0171]
应用实施例6
[0172]
取苯丙炔酸(0.5mmol)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基甲磺酰胺(0.55mmol)溶于3ml二氯甲烷中,在室温下搅拌反应,tlc监测反应过程,待原料酸消耗完全,减压蒸馏除去二氯甲烷,加入3ml乙腈作为溶剂,加入2-萘酚(0.55mmol)、三乙胺(0.05mmol),tlc监测反应过程,反应结束后在反应体系中加入0.5m的稀盐酸10ml,搅拌10min,产物析出,过滤水洗,收集固体,得到纯的产物,白色固体,产率96%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0173][0174]1h nmr(400mhz,cdcl3)δ7.84(dd,j=20.9,9.9hz,3h),7.70
–
7.58(m,3h),7.48(dt,j=14.2,7.4hz,3h),7.38(t,j=7.5hz,2h),7.32(d,j=8.8hz,1h).
[0175]
13
c nmr(100mhz,cdcl3)δ152.57,147.92,133.81,133.31,131.81,131.17,129.74,128.79,127.92,127.89,126.86,126.14,120.84,119.39,118.74,88.95,80.46.
[0176]
hrms(esi)m/z calcd.for c
19
h
13
o2[m+h]
+
:273.0910,found:273.0915。
[0177]
应用实施例7
[0178]
取喹啉-2-甲酸(0.5mmol)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基甲磺酰胺(0.55mmol)溶于3ml二氯甲烷中,在室温下搅拌反应,tlc监测反应过程,待原料酸消耗完全,减压蒸馏除去二氯甲烷,加入3ml乙腈作为溶剂,加入正丙醇(2.5mmol)、三乙胺(0.2mmol),tlc监测反应过程,反应结束后加入15ml乙酸乙酯稀释反应液,用0.5m的稀盐酸15ml震摇洗涤有机相2次,分去水相,用无水硫酸镁干燥有机相,减压蒸馏除去溶剂,得到纯的产物,淡黄色固体,产率93%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0179][0180]1hnmr(400mhz,cdcl3)δ8.31(dd,j=10.3,8.9hz,2h),8.17(d,j=8.5hz,1h),7.87(d,j=8.2hz,1h),7.78(ddd,j=8.4,6.9,1.4hz,1h),7.64(ddd,j=8.0,7.0,1.1hz,1h),4.46(t,j=6.9hz,2h),1.94
–
1.86(m,2h),1.06(t,j=7.4hz,3h).
[0181]
13
c nmr(100mhz,cdcl3)δ165.52,148.40,147.77,137.28,130.92,130.27,129.38,128.61,127.59,121.10,67.78,22.21,10.49.
[0182]
hrms(esi)m/z calcd.for c
13
h
14
no2[m+h]
+
:216.1019,found:216.1024。
[0183]
应用实施例8
[0184]
取cbz-phe-oh(0.5mmol)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基甲磺酰胺(0.55mmol)溶于3ml二氯甲烷中,在室温下搅拌反应,tlc监测反应过程,待原料酸消耗完全,减压蒸馏除去二氯甲烷,加入3ml乙腈作为溶剂,加入4-甲氧基苯酚(0.55mmol)、n,n-二异丙基乙胺(0.05mmol),tlc监测反应过程,在反应体系中加入0.5m的稀盐酸10ml,搅拌10min,产物析出,过滤水洗,收集固体,得到纯的产物,白色固体,产率94%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0185][0186]1h nmr(400mhz,cdcl3)δ7.33
–
7.19(m,10h),6.91
–
6.81(m,4h),5.38(s,1h),5.11(s,2h),4.87(q,j=5.7hz,1h),3.76(s,3h),3.23(d,j=5.7hz,2h).
[0187]
13
c nmr(100mhz,cdcl3)δ170.63,157.55,155.78,143.85,136.28,135.61,
129.53,128.81,128.62,128.29,128.18,127.40,122.08,114.56,67.16,55.65,55.08,38.37.
[0188]
hrms(esi)m/z calcd.for c
24
h
24
no5[m+h]
+
:406.1649,found:406.1653.ee>99%。
[0189]
应用实施例9
[0190]
取对硝基苯甲酸(0.5mmol)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基甲磺酰胺(0.55mmol)溶于3ml二氯甲烷中,在室温下搅拌反应,tlc监测反应过程,待原料酸消耗完全,减压蒸馏除去二氯甲烷,加入3ml乙腈作为溶剂,加入苄硫醇(0.55mmol)、n,n-二异丙基乙胺(0.05mmol),tlc监测反应过程,在反应体系中加入0.5m的稀盐酸10ml,搅拌10min,产物析出,过滤水洗,收集固体,得到纯的产物,白色固体,产率90%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0191][0192]1h nmr(400mhz,cdcl3)δ8.28(d,j=8.9hz,2h),8.10(d,j=8.9hz,2h),7.32(ddt,j=22.8,14.6,7.4hz,5h),4.36(s,2h).
[0193]
13
c nmr(100mhz,cdcl3)δ189.89,150.69,141.52,136.72,129.12,128.91,128.42,127.79,124.01,34.02.
[0194]
hrms(esi)m/z calcd.for c
14
h
12
no3s[m+h]
+
:274.0532,found:274.0538。
[0195]
应用实施例10
[0196]
取boc-ala-oh(0.5mmol)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基甲磺酰胺(0.55mmol)溶于3ml二氯甲烷中,在室温下搅拌反应,tlc监测反应过程,待原料酸消耗完全,减压蒸馏除去二氯甲烷,加入3ml乙腈作为溶剂,加入2-萘硫酚(0.55mmol)、n,n-二异丙基乙胺(0.05mmol),tlc监测反应过程,反应结束,在反应体系中加入0.2m的稀盐酸20ml,搅拌10min,产物析出,过滤水洗,收集固体,得到纯的产物,白色固体,产率94%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0197][0198]1h nmr(400mhz,cdcl3)δ7.94(s,1h),7.87
–
7.79(m,3h),7.54
–
7.47(m,2h),7.43(d,j=8.3hz,1h),5.10(s,1h),4.65
–
4.43(m,1h),1.50(s,9h),1.46(d,j=7.2hz,3h).
[0199]
13
c nmr(100mhz,cdcl3)δ200.34,155.08,134.70,133.70,133.48,131.10,128.91,128.04,127.90,127.25,126.66,124.75,80.56,56.47,28.52,18.83.
[0200]
hrms(esi)m/z calcd.for c
18
h
22
no3s[m+h]
+
:332.1315,found:332.1319.ee>99%。
[0201]
应用实施例11
[0202]
取4-硝基苯甲酸(0.5mmol)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基苯磺酰胺(0.55mmol)溶于3ml二氯甲烷中,在室温下搅拌反应,tlc监测反应过程,待原料酸消耗完全,减压蒸馏除去二氯甲烷,加入3ml乙腈作为溶剂,加入三氟乙醇(0.55mmol)、三乙
胺(0.05mmol),tlc监测反应过程,反应结束后加入15ml乙酸乙酯稀释反应液,用0.5m的稀盐酸15ml震摇洗涤有机相2次,分去水相,用无水硫酸镁干燥有机相,减压蒸馏除去溶剂,得到纯的产物,白色固体,产率95%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0203][0204]1h nmr(400mhz,cdcl3)δ8.34(d,j=6.9hz,2h),8.26(d,j=9.0hz,2h),4.76(q,j=8.3hz,2h).
[0205]
13
c nmr(100mhz,cdcl3)δ163.29,151.25,133.82,131.32,123.91,62.10,61.73,61.36,60.99.
[0206]
hrms(esi)m/z calcd.for c9h6f3nnao4[m+na]
+
:272.0141,found:272.0135.
[0207]
应用实施例12
[0208]
取boc-ala-oh(0.5mmol)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基苯磺酰胺(0.55mmol)溶于3ml二氯甲烷中,在室温下搅拌反应,tlc监测反应过程,待原料酸消耗完全,减压蒸馏除去二氯甲烷,加入3ml乙腈作为溶剂,加入2-萘硫酚(0.55mmol)、n,n-二异丙基乙胺(0.05mmol),tlc监测反应过程,反应结束,在反应体系中加入0.2m的稀盐酸20ml,搅拌10min,产物析出,过滤水洗,收集固体,得到纯的产物,白色固体,产率94%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0209][0210]1h nmr(400mhz,cdcl3)δ7.94(s,1h),7.87
–
7.79(m,3h),7.54
–
7.47(m,2h),7.43(d,j=8.3hz,1h),5.10(s,1h),4.65
–
4.43(m,1h),1.50(s,9h),1.46(d,j=7.2hz,3h).
[0211]
13
c nmr(100mhz,cdcl3)δ200.34,155.08,134.70,133.70,133.48,131.10,128.91,128.04,127.90,127.25,126.66,124.75,80.56,56.47,28.52,18.83.
[0212]
hrms(esi)m/z calcd.for c
18
h
22
no3s[m+h]
+
:332.1315,found:332.1319.ee>99%。
[0213]
应用实施例13
[0214]
取吡啶-2-甲酸(0.5mmol)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基苯磺酰胺(0.55mmol)溶于3ml二氯甲烷中,在室温下搅拌反应,tlc监测反应过程,待原料酸消耗完全,加入叔丁胺(0.55mmol),tlc监测反应过程,反应结束后加入15ml乙酸乙酯稀释反应液,用0.5m的稀盐酸15ml震摇洗涤有机相2次,分去水相,用无水硫酸镁干燥有机相,减压蒸馏除去溶剂,得到纯的产物,白色固体,产率97%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0215]
[0216]1h nmr(400mhz,cdcl3)δ8.52(d,j=4.7hz,1h),8.18(d,j=7.8hz,1h),8.01(s,1h),7.83(td,j=7.7,1.5hz,1h),7.40(dd,j=7.5,4.8hz,1h),1.50(s,9h).
[0217]
13
c nmr(100mhz,cdcl3)δ163.56,150.95,147.87,137.48,125.96,121.84,51.05,28.91.
[0218]
hrms(esi)m/z calcd.for c10h14n2nao[m+na]
+
:201.0998,found:201.1005.
[0219]
应用实施例14
[0220]
取cbz-ala-oh(0.5mmol)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基苯磺酰胺(0.55mmol)溶于5ml二氯甲烷中,在室温下搅拌反应,tlc监测反应过程,待原料酸消耗完全,加入h-phe-otbu(0.55mmol),tlc监测反应过程,反应结束后加入15ml乙酸乙酯稀释反应液,用0.2m的稀盐酸15ml震摇洗涤有机相3次,分去水相,用无水硫酸镁干燥有机相,浓缩后用二氯甲烷/石油醚重结晶,得到纯的产物,白色固体,产率93%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0221][0222]1h nmr(400mhz,cdcl3)δ7.38
–
7.28(m,5h),7.26
–
7.19(m,3h),7.13(d,j=6.8hz,2h),6.69
–
6.45(m,1h),5.51
–
5.34(m,1h),5.10(t,j=9.4hz,2h),4.72(q,j=6.2hz,1h),4.34
–
4.16(m,1h),3.11
–
3.03(m,2h),1.40(s,9h),1.33(d,j=6.8hz,3h).
[0223]
13
c nmr(100mhz,cdcl3)δ171.82,170.36,155.93,136.35,136.14,129.60,128.61,128.45,128.25,128.12,127.05,82.52,67.07,53.72,50.56,38.06,28.04,18.69.
[0224]
hrms(esi)m/z calcd.for c
24
h
31
n2o5[m+h]
+
:427.2227,found:427.2235.dr>99:1。
[0225]
应用实施例15
[0226]
取喹啉-2-甲酸(0.5mmol)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基对甲苯磺酰胺(0.55mmol)溶于3ml二氯甲烷中,在室温下搅拌反应,tlc监测反应过程,待原料酸消耗完全,减压蒸馏除去二氯甲烷,加入3ml乙腈作为溶剂,加入正丙醇(2.5mmol)、三乙胺(0.2mmol),tlc监测反应过程,反应结束后加入15ml乙酸乙酯稀释反应液,用0.5m的稀盐酸15ml震摇洗涤有机相2次,分去水相,用无水硫酸镁干燥有机相,减压蒸馏除去溶剂,得到纯的产物,淡黄色固体,产率91%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0227][0228]1hnmr(400mhz,cdcl3)δ8.31(dd,j=10.3,8.9hz,2h),8.17(d,j=8.5hz,1h),7.87(d,j=8.2hz,1h),7.78(ddd,j=8.4,6.9,1.4hz,1h),7.64(ddd,j=8.0,7.0,1.1hz,1h),4.46(t,j=6.9hz,2h),1.94
–
1.86(m,2h),1.06(t,j=7.4hz,3h).
[0229]
13
c nmr(100mhz,cdcl3)δ165.52,148.40,147.77,137.28,130.92,130.27,129.38,128.61,127.59,121.10,67.78,22.21,10.49.
[0230]
hrms(esi)m/z calcd.for c
13
h
14
no2[m+h]
+
:216.1019,found:216.1024。
[0231]
应用实施例16
[0232]
取对硝基苯甲酸(0.5mmol)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基对甲苯磺酰胺(0.55mmol)溶于3ml二氯甲烷中,在室温下搅拌反应,tlc监测反应过程,待原料酸消耗完全,减压蒸馏除去二氯甲烷,加入3ml乙腈作为溶剂,加入苄硫醇(0.55mmol)、n,n-二异丙基乙胺(0.05mmol),tlc监测反应过程,在反应体系中加入0.5m的稀盐酸10ml,搅拌10min,产物析出,过滤水洗,收集固体,得到纯的产物,白色固体,产率88%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0233][0234]1h nmr(400mhz,cdcl3)δ8.28(d,j=8.9hz,2h),8.10(d,j=8.9hz,2h),7.32(ddt,j=22.8,14.6,7.4hz,5h),4.36(s,2h).
[0235]
13
c nmr(100mhz,cdcl3)δ189.89,150.69,141.52,136.72,129.12,128.91,128.42,127.79,124.01,34.02.
[0236]
hrms(esi)m/z calcd.for c
14
h
12
no3s[m+h]
+
:274.0532,found:274.0538。
[0237]
应用实施例17
[0238]
取苯并噻吩-2-甲酸(0.5mmol)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基对甲苯磺酰胺(0.55mmol)溶于3ml二氯甲烷中,在室温下搅拌反应,tlc监测反应过程,待苯丙炔酸消耗完全,加入2-羟基乙胺(0.55mmol),tlc监测反应过程,反应结束后加入15ml乙酸乙酯稀释反应液,用0.5m的稀盐酸15ml震摇洗涤有机相2次,分去水相,用无水硫酸镁干燥有机相,减压蒸馏除去溶剂,得到纯的产物,白色固体,产率94%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0239][0240]1h nmr(400mhz,cdcl3)δ7.84(dd,j=5.9,3.2hz,1h),7.80(dd,j=5.9,3.2hz,1h),7.44(s,1h),7.38(dt,j=6.0,3.5hz,2h),3.71
–
3.64(m,4h),1.73
–
1.62(m,6h).
[0241]
13
c nmr(100mhz,cdcl3)δ=163.8,140.2,138.8,137.4,125.6,124.8,124.7,124.6,122.4,26.3,24.7.
[0242]
hrms(esi)m/z calcd.for c
14
h
16
nos[m+h]
+
:246.0947,found:246.0953。
[0243]
应用实施例18
[0244]
取boc-ala-oh(0.5mmol)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基对甲苯磺酰胺(0.55mmol)溶于5ml二氯甲烷中,在室温下搅拌反应,tlc监测反应过程,待原料酸消耗完全,加入h-phe-otbu(0.55mmol),tlc监测反应过程,反应结束后加入15ml乙酸乙酯稀释反应液,用0.2m的稀盐酸15ml震摇洗涤有机相3次,分去水相,用无水硫酸镁干燥有机相,浓缩后用二氯甲烷/石油醚重结晶,得到纯的产物,白色固体,产率97%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0245][0246]1h nmr(400mhz,cdcl3)δ7.25(dt,j=13.1,6.8hz,3h),7.15(d,j=6.8hz,2h),6.60(s,1h),5.05(s,1h),4.71(q,j=6.2hz,1h),4.15(s,1h),3.13
–
3.04(m,2h),1.43(s,9h),1.39(s,9h),1.32(d,j=7.0hz,3h).
[0247]
13
c nmr(100mhz,cdcl3)δ172.23,170.41,155.40,136.24,129.64,128.44,127.02,82.44,80.13,53.71,50.28,38.16,28.40,28.03,18.55.
[0248]
hrms(esi)m/z calcd.for c
21
h
33
n2o5[m+h]
+
:393.2384,found:393.2389.dr>99:1。
[0249]
应用实施例19
[0250]
取cbz-gly-oh(0.5mmol)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基三氟乙酰胺(0.55mmol)溶于3ml二氯甲烷中,在室温下搅拌反应,tlc监测反应过程,待原料酸消耗完全,减压蒸馏除去二氯甲烷,加入3ml乙腈作为溶剂,加入雌酚酮(0.55mmol)、n,n-二异丙基乙胺(0.05mmol),tlc监测反应过程,反应结束,在反应体系中加入0.2m的稀盐酸20ml,搅拌10min,产物析出,过滤水洗,收集固体,得到纯的产物,白色固体,产率90%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0251][0252]1h nmr(400mhz,cdcl3)δ7.38
–
7.25(m,6h),6.91
–
6.72(m,2h),5.46(s,1h),5.14(s,2h),4.20(d,j=5.5hz,2h),2.94
–
2.84(m,2h),2.50(dd,j=18.9,8.7hz,1h),2.42
–
2.34(m,1h),2.30
–
2.22(m,1h),2.18
–
1.94(m,4h),1.65
–
1.41(m,6h),0.90(s,3h).
[0253]
13
c nmr(100mhz,cdcl3)δ168.95,156.37,148.18,138.18,137.80,136.20,128.56,128.23,128.10,126.47,121.28,118.43,67.21,50.43,47.93,44.14,42.99,37.98,35.84,31.56,29.38,26.31,25.76,21.59,13.83.
[0254]
hrms(esi)m/z calcd.for c
28
h
32
no5[m+h]
+
:462.2275,found:462.2281。
[0255]
应用实施例20
[0256]
取吡啶-2-甲酸(0.5mmol)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基三氟乙酰胺(0.55mmol)溶于3ml二氯甲烷中,在室温下搅拌反应,tlc监测反应过程,待原料酸消耗完全,减压蒸馏除去二氯甲烷,加入3ml乙腈作为溶剂,加入苄硫醇(0.55mmol)、三乙胺(0.05mmol),tlc监测反应过程,在反应体系中加入0.5m的稀盐酸10ml,搅拌10min,产物析出,过滤水洗,收集固体,得到纯的产物,白色固体,产率89%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0257]
[0258]1h nmr(400mhz,cdcl3)δ8.66(d,j=4.7hz,1h),7.96(d,j=7.8hz,1h),7.83(td,j=7.7,1.6hz,1h),7.50
–
7.46(m,1h),7.39(d,j=7.2hz,2h),7.29(t,j=7.4hz,2h),7.26
–
7.21(m,1h),4.28(s,2h).
[0259]
13
c nmr(100mhz,cdcl3)δ193.07,151.99,149.23,137.65,137.31,129.12,128.66,127.93,127.28,120.56,33.29.
[0260]
hrms(esi)m/z calcd.for c
13
h
12
nos[m+h]
+
:230.0634,found:230.0640。
[0261]
应用实施例21
[0262]
取肉桂酸(0.5mmol)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基三氟乙酰胺(0.55mmol)溶于3ml二氯甲烷中,在室温下搅拌反应,tlc监测反应过程,待原料酸消耗完全,加入2-苯乙胺(0.55mmol),tlc监测反应过程,反应结束后加入15ml乙酸乙酯稀释反应液,用0.5m的稀盐酸15ml震摇洗涤有机相2次,分去水相,用无水硫酸镁干燥有机相,减压蒸馏除去溶剂,得到纯的产物,白色固体,产率91%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0263][0264]1h nmr(400mhz,cdcl3)δ7.61(d,j=15.6hz,1h),7.46(dd,j=6.4,2.5hz,2h),7.37
–
7.28(m,5h),7.23(t,j=9.5hz,3h),6.35(d,j=15.6hz,1h),5.86(s,1h),3.65(q,j=6.8hz,2h),2.88(t,j=6.9hz,2h).
[0265]
13
c nmr(100mhz,cdcl3)δ166.04,141.08,139.00,134.95,129.74,128.90,128.89,128.78,127.88,126.65,120.83,40.96,35.80.
[0266]
hrms(esi)m/z calcd.for c
17
h
18
no[m+h]
+
:252.1383,found:252.1390。
[0267]
应用实施例22
[0268]
取fmoc-ser(tbu)-oh(0.5mmol)、n-(2,2-二甲基-1,3-二氧戊环-4-基甲基)-n-乙炔基三氟乙酰胺(0.55mmol)溶于5ml二氯甲烷中,在室温下搅拌反应,tlc监测反应过程,待原料酸消耗完全,加入h-leu-otbu(0.55mmol),tlc监测反应过程,反应结束后加入15ml乙酸乙酯稀释反应液,用0.2m的稀盐酸15ml震摇洗涤有机相3次,分去水相,用无水硫酸镁干燥有机相,浓缩后用二氯甲烷/石油醚重结晶,得到纯的产物,白色固体,产率96%。以下是产物的结构式及核磁共振实验数据、质谱实验数据:
[0269][0270]1h nmr(400mhz,cdcl3)δ7.75(d,j=7.5hz,2h),7.60(d,j=6.9hz,2h),7.39(t,j=7.4hz,2h),7.30(t,j=7.4hz,2h),7.22(s,1h),5.80(s,1h),4.49(s,1h),4.42
–
4.36(m,2h),4.23(t,j=7.1hz,2h),3.83(dd,j=8.3,3.4hz,1h),3.40(t,j=8.0hz,1h),1.76
–
1.59(m,3h),1.58
–
1.47(m,3h),1.45(s,9h),1.22(s,9h),0.95(d,j=6.2hz,6h).
[0271]
13
c nmr(100mhz,cdcl3)δ171.69,169.99,156.11,143.98,143.86,141.36,127.77,127.14,125.20,120.03,81.76,74.31,67.21,61.89,54.41,51.74,47.25,41.96,
28.06,27.43,24.95,22.90,22.24.
[0272]
hrms(esi)m/z calcd.for c
32
h
45
n2o6[m+h]
+
:553.3272,found:553.3279.dr>99:1。
[0273]
以上所述的实施例仅仅是对本发明的优选实施方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案作出的各种变形和改进,均应落入本发明权利要求书确定的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1