一种比拉斯汀中间体化合物的制作方法
1.本发明属于药物合成技术领域,具体涉及一种比拉斯汀中间体化合物。
背景技术:
2.比拉斯汀(bilastine),化学名为4-[2-[4-[1-(2-乙氧基乙基)-1h-2-苯并咪唑基]-1-哌啶基]-乙基]-α,α-二甲基苯乙酸,cas号202189-78-4,是西班牙faes制药公司开发的一种口服第2代非镇静组胺h1受体拮抗药,2012年8月被欧盟批准用于治疗过敏性鼻炎和荨麻疹。本品选择性作用于外周组胺受体,对其他组胺受体无影响,无心脏毒性,口服给药吸收迅速,具有良好的耐受性、安全性和较高的生物利用度。其化学结构式为:
[0003][0004]
目前比拉斯汀的合成方法主要有以下几种:
[0005]
①
专利ep0818454、ep0580541、us5877187、cn1176964a、cn109694367a、cn1105716c、es2151442、es2151442a1、cn104402773a及cn103351380a以4-溴苯乙酸甲酯或其下游中间体为原料,经甲基化、水解、羧基保护和格氏反应得到4-[2-甲基-2-(4,5-二氢-4,4-二甲基噁唑-2-基)乙基]苯乙醇,羟基经离去基团(如cl、br、i、磺酸酯等)取代后,先后经2-(4-哌啶基)-1h-苯并咪唑、2-氯乙基乙醚取代,最后水解得到目标产品。反应路线如下:
[0006][0007]
但该路线存在以下问题:
①
反应中用到丁基锂或格式试剂,对基团兼容性较差,需要无水无氧,条件要求苛刻;
②
同时环氧乙烷是危险品,所用甲基化试剂碘甲烷沸点低、毒性均较大,操作安全性较低;
③
并且需要用到较特殊的噁唑环来保护羧基,反应条件苛刻;
④
整体路线较长,收率低,不适合工业化生产。
[0008]
②
中国专利cn104326909a及文献比拉斯汀重要中间体的合成,《中国医药工业杂志》,2015,46(7):677-679以α,α-二甲基苯乙酸酯为起始物料,先经傅-克酰基化和还原反应制得α,α-二甲基-4-(2-溴乙基)苯乙酸甲酯,依次与2-(4-哌啶基)-1h-苯并咪唑和2-氯乙基乙醚发生亲核取代反应后,水解得比拉斯汀。反应路线如下:
[0009][0010]
另外,以上路线中均以2-(4-哌啶基)苯并咪唑为原料,分子结构中含有两个易于被取代的氢,对选择性要求较高,反应条件苛刻,且容易产生副产物。
[0011]
③
中国专利cn102675101a以α,α-二甲基苯乙酸酯为原料,经傅-克酰基化和还原反应制得α,α-二甲基-4-(2-卤乙基)苯乙酸酯,再与1-(2-乙氧基乙基)-2-(4-哌啶基)-1h-苯并咪唑经取代、水解等反应制得目标产品。反应路线如下:
[0012][0013]
但该法采用wolff-kishner-黄鸣龙法还原羰基,其反应条件需高温、毒性较大;且1-(2-乙氧基乙基)-2-(4-哌啶基)-1h-苯并咪唑的合成需要经过上保护基、取代、脱保护基等反应,步骤繁琐、不适合工业化生产。
[0014]
④
文献j.org.chem.,1988,53(6):1170-1176、synth.commun.,2011,41(9):1394-1402及比拉斯汀关键中间体的合成,《河北化工》,2013,36(3):14-15以α,α-二甲基-4-溴苯乙酸甲酯为原料,通过stille偶联反应,再经水合、上保护基、烷基化、脱保护基和酯水解等反应制得比拉斯汀。但该方法使用了对环境不友好的有机锡、硼烷二甲硫醚络合物,后处理成本高。反应路线如下:
[0015][0016]
⑤
专利wo2009102155(cn101952273a)以4-溴苯乙醇为原料经钯催化偶联、磺酰化,再与1-(2-乙氧基乙基)-2-(4-哌啶基)-1h-苯并咪唑经取代、水解等反应制得目标产品。但是该方法中,关键中间体2-(4-羟乙基苯基)-2-甲基丙酸乙酯的合成是以对溴苯乙醇与1-甲氧基-1-(三甲基甲硅氧基)-2-甲基-1-丙烯为原料,在催化剂双(二亚芐基丙酮)钯,三叔丁基磷和氟化锌存在下反应制得。反应路线如下:
[0017][0018]
该反应主要有三个缺点:
①
原料1-甲氧基-1-(三甲基甲硅氧基)-2-甲基-1-丙烯及催化剂双(二亚芐基丙酮)钯和三叔丁基磷等价格都极其昂贵,不易购得,且不易保存;
②
该反应需要极为严格的无水无氧条件,操作复杂,且所得产物很难纯化;
③
反应完成后剩余
的钯和磷都会对环境造成严重污染。
[0019]
⑥
中国专利cn106146459a及文献比拉斯汀重要中间体的合成,《中国医药工业杂志》,2016,47(11):1363-1365则采用廉价易得的2-硝基苯胺为原料,先与4-甲酰基哌啶-1-羧酸叔丁酯经还原-关环反应制得4-(1h-苯并[d]咪唑-2-基)哌啶-1-甲酸叔丁酯,然后再与氯乙基乙醚发生n-烷基化反应、水解反应,最后与2-甲基-2-(4-(2-(甲苯磺酰氧基)乙基)苯基)丙酸钠经取代反应制得比拉斯汀。反应路线如下:
[0020][0021]
但该反应路线中化合物4-甲酰基哌啶-1-羧酸叔丁酯和2-甲基-2-(4-(2-(甲苯磺酰氧基)乙基)苯基)丙酸钠均不能从市场购得,因此反应路线较长,另外在比拉斯汀合成过程中,需要进行20小时的纯化,大大延长了工艺时间,降低了工业化生产效率。
[0022]
⑦
中国专利cn110903278a以哌啶-4-羧酸为起始物料,经α,α-二甲基-4-(2-溴乙基)苯乙酸甲酯取代后,与邻苯二胺环化制得α,α-二甲基-4-(2-(4-(1h-2-苯并[d]咪唑基)哌啶-1-基)乙基)苯乙酸甲酯,然后与氯乙基乙醚发生n-烷基化反应、水解反应制得比拉斯汀。反应路线如下:
[0023][0024]
但是该工艺中的起始物料α,α-二甲基-4-(2-溴乙基)苯乙酸甲酯需要参照文献比拉斯汀重要中间体的合成,《中国医药工业杂志》,2015,46(7):677-679中的方法制备(α,α-二甲基-苯乙酸甲酯与溴乙酰溴先经傅-克酰基化反应后、再经三氟乙酸/三乙基硅烷体系还原制得),不仅延长了反应步骤,同时傅-克酰基化反应用到活性较高的alcl3催化,其用量大,后处理不仅危险,同时产生的大量铝盐影响产品的分离,此外还原体系运行成本较高。同时环化步骤中用到大量缩合剂,如:二环己基碳二亚胺、n-羟基琥珀酰亚胺、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、1-羟基苯并三唑等,不仅原子经济性较差,同时后处理同样繁琐,较难实现产业化放大生产。
[0025]
⑧
中国专利cn111039922a以2-(4-(2-羟乙基)苯基)-2-甲基丙酸为一起始物料,与碘甲烷反应后生成2-(4-(2-碘乙基)苯基)-2-甲基丙酸甲酯,以4-(1-(2-乙氧基乙基)-1h-苯并[d]咪唑-2-基)哌啶-1-甲酸叔丁酯为另一起始物料,先脱除boc保护基,再与2-(4-(2-碘乙基)苯基)-2-甲基丙酸甲酯反应后,经酯水解制得目标产品。但是该工艺同样用到毒性及沸点较低的碘甲烷进行碘代反应,操作安全性较低。反应路线如下:
[0026][0027]
⑨
中国专利cn110950837a及cn107365297a则以4-羟乙基苯基叔丁酸酯或其下游中间体为起始物料,先通过氧化反应生成4-乙醛基苯基叔丁酸酯,然后与1-(2-乙氧基乙基)-2-(哌啶-4-基)-1h-苯并[d]咪唑经nabh4或libh4还原胺化后再水解制得比拉斯汀。但氧化反应在工业化中操作较危险,同时也容易得到过氧化的酸杂质。反应路线如下:
[0028]
⑩
中国专利cn104530002a及cn104177331a以对甲基苯乙醇或其下游中间体为起始原料,经对甲苯磺酰氯磺酰化后制得磺酸酯,然后与1-乙氧基乙基-2-哌啶基苯并咪唑反应,再在苄位溴化,经格氏反应在苄位引入羧基后将羧基转化成甲酯,再以硫酸二甲酯或碘甲烷最后上甲基后再水解在苄位进行二甲基化,最后进行水解反应制得比拉斯汀。反应中用到格式试剂引入羧基,需要无水无氧操作,条件要求苛刻,工业化难度较大;反应路线如下:
[0029][0030]
鉴于目前比拉斯汀的制备工艺中存在以上较多不足。因此,研究寻找一条反应条件温和,操作过程简便,产品收率高、纯度高的适合工业化生产比拉斯汀的工艺仍是目前需要解决的问题。
技术实现要素:
[0031]
针对目前现有比拉斯汀制备技术存在的问题,本发明提供了一种新的比拉斯汀中间体化合物及其制备方法,同时提供了利用该新中间体制备比拉斯汀的方法。该方法反应条件温和,操作过程简便,所制得的目标产品具有较高的纯度、收率。
[0032]
本发明的具体技术方案如下:
[0033]
本发明第一方面提供了一种比拉斯汀中间体化合物,其结构如式i-1所示:
[0034][0035]
本发明第二方面提供了一种比拉斯汀中间体化合物i-1的制备方法,具体包括以下步骤:室温,将sm-1、缚酸剂加入反应溶剂中,加入(e)-1,2-二溴乙烯,控温至反应结束后,经后处理制得中间体i-1。
[0036][0037]
优选地,所述的反应溶剂选自四氢呋喃、乙腈、1,4-二氧六环,n,n-二甲基甲酰胺中的一种或其组合,其中特别优选n,n-二甲基甲酰胺。
[0038]
优选地,所述的缚酸剂选自三乙胺、n,n-二异丙基乙基胺、碳酸钾、碳酸钠中的一种或其组合,其中特别优选三乙胺。
[0039]
优选地,所述的化合物sm-1与缚酸剂、化合物sm-2的投料摩尔比为1:1.6~3.0:1.0~1.5,其中特别优选1:2.2:1.05。
[0040]
优选地,所述的反应温度为60~110℃,其中特别优选80~85℃。
[0041]
优选方案,所述的后处理步骤为:将反应液降温至室温,过滤,滤液加入纯化水中,二氯甲烷提取,合并有机相,有机相经饱和食盐水洗,无水硫酸钠干燥,过滤,滤液减压浓缩至干即为中间体化合物i-1。
[0042]
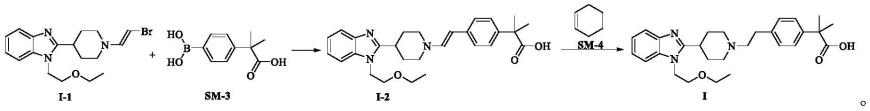
本发明第三方面提供了一种化合物i-1制备比拉斯汀的用途。
[0043]
化合物i-1用于制备比拉斯汀的方法,包括如下方案:
[0044]
化合物i-1在催化剂的作用下与2-(4-硼苯基)-2-甲基丙酸反应得化合物i-2;化合物i-2还原得比拉斯汀,合成路线如下:
[0045][0046]
优选地,在以下部分将详细介绍以上步骤:
[0047]
化合物i-2的制备
[0048]
化合物i-2的制备方法具体包括如下步骤:惰性气体保护,室温条件下,将催化剂加入反应溶剂b中,搅拌混匀后,向反应溶剂中加入碱及纯化水,加入化合物i-1与化合物sm-3,控温至反应结束后,反应经后处理得中间体i-2。
[0049]
优选地,所述的催化剂选自pd(pph3)4、pd(pph3)2cl2、pd(dppf)cl2中的一种或其组合,特别优选pd(pph3)2cl2。
[0050]
优选地,所述的反应溶剂b选自1,4-二氧六环、甲苯、n,n-二甲基甲酰胺、二甲基亚砜中的一种或其组合,特别优选二甲基亚砜。
[0051]
优选地,所述的碱选自k2co3、na2co3、k3po4、na3po4、naoac、koac中的一种,特别优选k3po4。
[0052]
优选地,所述的化合物i-1与化合物sm-3、碱、催化剂的投料摩尔比为1:1.0~2.0:2.0~3.5:0.03~0.08,特别优选1:1.2:2.8:0.05。
[0053]
优选地,所述的反应温度为80~110℃。
[0054]
优选地,所述的惰性气体为氩气、氮气中的一种或其组合,其中特别优选氩气。
[0055]
在一优选方案中,后处理步骤为控温至反应结束后,过滤,将滤液加入纯化水中,萃取剂提取,合并有机相,无水硫酸钠干燥,过滤,滤液减压浓缩至干后制得中间体i-2;优选地,所述的萃取剂为二氯甲烷、氯仿、乙酸乙酯、甲基叔丁基醚中的一种或其组合,特别优选乙酸乙酯。
[0056]
比拉斯汀i的制备
[0057]
比拉斯汀的制备方法具体包括如下步骤:室温下,将中间体i-2、环己烯、钯碳加入反应溶剂c中,控温至反应结束后,反应经后处理得目标产品比拉斯汀i。
[0058]
优选地,所述的反应溶剂c选自甲苯、二甲苯中的一种或其组合,其中特别优选甲苯。
[0059]
优选地,所述的化合物i-2与环己烯的投料摩尔比为1:1.4~2.6,其中特别优选1:1.8。
[0060]
优选地,所述的化合物i-2与钯碳的投料质量比为1:0.05~0.15,其中特别优选1:0.1。
[0061]
优选地,所述的反应温度为90~130℃,其中特别优选105~110℃。
[0062]
优选地,所述的后处理步骤为:将反应液降温至室温,过滤,滤液经纯化水洗,饱和食盐水洗,无水硫酸钠干燥,过滤,滤液减压浓缩至干即为比拉斯汀i。
[0063]
本发明的有益效果:
[0064]
1.本发明提供了一种新的比拉斯汀中间体化合物及其制备方法,该新中间体以1-(2-乙氧基乙基)-2-(哌啶-4-基)-1h-苯并[d]咪唑(sm-1)为起始物料,经(e)-1,2-二溴乙烯取代即得新中间体化合物,合成方法简单,该新中间体在后续取代反应中无较大杂质产生。
[0065]
2.本发明所得新中间体与2-(4-硼苯基)-2-甲基丙酸经suzuki偶联后还原得吡拉斯汀本发明的比拉斯汀的制备工艺,相较于现有技术所得产品具有较高的收率与纯度,并且操作简便、安全,适合工业化生产。
具体实施方式
[0066]
下面通过实施例来进一步说明本发明,应该正确理解的是:本发明的实施例仅仅是用于说明本发明,而不是对本发明的限制,所以,在本发明的方法前提下对本发明的简单改进均属于本发明要求保护的范围。
[0067]
对本发明得到的中间体及产物结构确证数据如下:
[0068][0069]
esi-hrms(m/z):380.1158[m+h]
+
;1h nmr(400mhz,dmso-d6)δ7.56~7.50(m,1h),7.48~7.43(m,1h),7.20~7.09(m,2h),6.52(d,j=15.8hz,1h),5.07(d,j=15.8hz,1h),4.46(t,j=6.9hz,2h),3.80(t,j=6.9hz,2h),3.66~3.40(m,4h),3.11~2.92(m,1h),2.76~2.56(m,2h),2.30~2.14(m,2h),2.06~1.90(m,2h),1.22(t,j=6.8hz,3h);
13
c nmr(101mhz,dmso-d6):δ162.35,145.80,138.62,136.48,123.50,122.51,119.86,111.81,78.95,70.18,66.68,48.50,46.37,29.15,27.91,14.84。
[0070][0071]
esi-hrms(m/z):460.5978[m+h]-;1h nmr(400mhz,dmso-d6)δ7.62~7.54(m,3h),7.52(d,j=7.6hz,2h),7.36(d,j=7.6hz,2h),7.22~7.14(m,2h),5.12(d,j=16.1hz,1h),4.45(t,j=6.6hz,2h),4.00~3.90(m,1h),3.82(t,j=6.6hz,2h),3.49(d,j=6.8hz,2h),3.45~3.33(m,2h),3.16~3.04(m,2h),2.34~2.20(m,2h),1.85~1.74(m,2h),1.66(s,6h),1.20(t,j=6.8hz,3h);
13
c nmr(101mhz,dmso-d6):δ182.78,162.30,145.25,138.80,138.17,136.51,131.41,128.68,126.33,123.52,122.53,119.98,112.81,95.68,71.18,66.87,48.48,47.69,46.35,29.14,27.90,22.80,14.87。
[0072][0073]
esi-hrms(m/z):464.2929[m+h]
+
;1h nmr(400mhz,cd3od)δ7.63~7.49(m,2h),7.39(d,j=8.1hz,2h),7.31~7.21(m,2h),7.18(d,j=8.1hz,2h),4.51(t,j=4.8hz,2h),3.77(t,j=4.8hz,2h),3.58(d,j=12.3hz,2h),3.43(q,j=6.9hz,3h),3.12~3.07(m,2h),2.96~2.82(m,4h),2.28~2.17(m,4h),1.51(s,6h),1.08(t,j=6.9hz,3h);
13
c nmr(101mhz,cd3od):δ183.3,158.6,147.9,147.5,135.4,134.7,132.6,132.5,130.6,130.1,129.8,129.8,118.1,116.7,71.1,70.5,55.6,50.1,49.8,49.3,34.7,33.4,31.4,29.6,29.5,22.1,20.9,17.9。
[0074]
本发明采用hplc测定比拉斯汀的纯度,色谱条件如下:
[0075]
色谱柱:ymc-triart c
18
柱(4.6mm
×
150mm,5μm)或效能相当的色谱柱;
[0076]
流动相a:10mmol/l磷酸氢二钾:乙腈:四氢呋喃(用磷酸调ph 7.0)(750:150:100);
[0077]
流动相b:10mmol/l磷酸氢二钾:乙腈:四氢呋喃(用磷酸调ph 8.0)(150:800:50);
[0078]
梯度洗脱(0-30min:a 100%—70%,30—50min:a 75%—0%,50—60min,0%—0%);
[0079]
柱温:50℃;
[0080]
检测波长:210nm;
[0081]
流速:0.8ml/min;
[0082]
进样量:10μl;
[0083]
其中,比拉斯汀的保留时间为17.7min左右。
[0084]
以下各实施例中,未详细描述的各种过程和方法是本领域中公知的常规方法。
[0085]
中间体i-1的合成
[0086]
实施例1
[0087]
室温,将1-(2-乙氧基乙基)-2-(哌啶-4-基)-1h-苯并[d]咪唑(sm-1,54.68g,0.20mol)、三乙胺(44.52g,0.44mol)加入n,n-二甲基甲酰胺(300ml)中,加入(e)-1,2-二溴乙烯(sm-2,39.03g,0.21mol)的n,n-二甲基甲酰胺(100ml)溶液,控温80~85℃反应,经检测反应完毕后,将反应液降温至室温,过滤,滤液加入纯化水(2000ml)中,二氯甲烷(600ml
×
3)提取,合并有机相,有机相用饱和食盐水(500ml
×
2)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干即为中间体i-1,收率88.6%,纯度99.62%。
[0088]
实施例2
[0089]
室温,将1-(2-乙氧基乙基)-2-(哌啶-4-基)-1h-苯并[d]咪唑(sm-1,54.68g,0.20mol)、n,n-二异丙基乙基胺(56.87g,0.44mol)加入n,n-二甲基甲酰胺(300ml)中,加入(e)-1,2-二溴乙烯(sm-2,37.17g,0.20mol)的n,n-二甲基甲酰胺(100ml)溶液,控温105~110℃反应,经检测反应完毕后,将反应液降温至室温,过滤,滤液加入纯化水(2000ml)中,二氯甲烷(600ml
×
3)提取,合并有机相,饱和食盐水(500ml
×
2)洗,无水硫酸钠干燥,过滤
滤液减压浓缩至干即为中间体i-1,收率84.3%,纯度99.23%。
[0090]
实施例3
[0091]
室温,将1-(2-乙氧基乙基)-2-(哌啶-4-基)-1h-苯并[d]咪唑(sm-1,54.68g,0.20mol)、碳酸钾(60.81g,0.44mol)加入四氢呋喃(300ml)中,加入(e)-1,2-二溴乙烯(sm-2,55.76g,0.30mol)的四氢呋喃(100ml)溶液,控温60℃~66℃反应,经检测反应完毕后,将反应液降温至室温,过滤,滤液加入纯化水(2000ml)中,二氯甲烷(600ml
×
3)提取,合并有机相,饱和食盐水(500ml
×
2)洗,无水硫酸钠干燥,过滤滤液减压浓缩至干即为中间体i-1,收率85.2%,纯度99.21%。
[0092]
实施例4
[0093]
室温,将1-(2-乙氧基乙基)-2-(哌啶-4-基)-1h-苯并[d]咪唑(sm-1,54.68g,0.20mol)、碳酸钠(46.64g,0.44mol)加入乙腈(300ml)中,加入(e)-1,2-二溴乙烯(sm-2,63.19g,0.34mol)的乙腈(100ml)溶液,控温55℃~60℃反应,经检测反应完毕后,将反应液降温至室温,过滤,滤液加入纯化水(2000ml)中,二氯甲烷(600ml
×
3)提取,合并有机相,饱和食盐水(500ml
×
2)洗,无水硫酸钠干燥,过滤滤液减压浓缩至干即为中间体i-1,收率80.1%,纯度98.81%。
[0094]
实施例5
[0095]
室温,将1-(2-乙氧基乙基)-2-(哌啶-4-基)-1h-苯并[d]咪唑(sm-1,54.68g,0.20mol)、三乙胺(32.38g,0.32mol)加入乙腈(300ml)中,加入(e)-1,2-二溴乙烯(sm-2,39.03g,0.21mol)的乙腈(100ml)溶液,控温80~85℃回流反应,经检测反应完毕后,将反应液降温至室温,过滤,滤液加入纯化水(2000ml)中,二氯甲烷(600ml
×
3)提取,合并有机相,饱和食盐水(500ml
×
2)洗,无水硫酸钠干燥,过滤滤液减压浓缩至干即为中间体i-1,收率84.0%,纯度99.32%。
[0096]
实施例6
[0097]
室温,将1-(2-乙氧基乙基)-2-(哌啶-4-基)-1h-苯并[d]咪唑(sm-1,54.68g,0.20mol)、三乙胺(60.71g,0.60mol)加入1,4-二氧六环(300ml)中,加入(e)-1,2-二溴乙烯(sm-2,39.03g,0.21mol)的1,4-二氧六环(100ml)溶液,控温90~95℃反应,经检测反应完毕后,将反应液降温至室温,过滤,滤液加入纯化水(2000ml)中,二氯甲烷(600ml
×
3)提取,合并有机相,饱和食盐水(500ml
×
2)洗,无水硫酸钠干燥,过滤滤液减压浓缩至干即为中间体i-1,收率84.8%,纯度99.18%。
[0098]
实施例7
[0099]
室温,将1-(2-乙氧基乙基)-2-(哌啶-4-基)-1h-苯并[d]咪唑(sm-1,54.68g,0.20mol)、三乙胺(28.33g,0.28mol)加入n,n-二甲基甲酰胺(300ml)中,加入(e)-1,2-二溴乙烯(sm-2,39.03g,0.21mol)的n,n-二甲基甲酰胺(100ml)溶液,控温110~115℃反应,经检测反应完毕后,将反应液降温至室温,过滤,滤液加入纯化水(2000ml)中,二氯甲烷(600ml
×
3)提取,合并有机相,饱和食盐水(500ml
×
2)洗,无水硫酸钠干燥,过滤滤液减压浓缩至干即为中间体i-1,收率80.8%,纯度98.92%。
[0100]
实施例8
[0101]
室温,将1-(2-乙氧基乙基)-2-(哌啶-4-基)-1h-苯并[d]咪唑(sm-1,54.68g,0.20mol)、三乙胺(64.76g,0.64mol)加入1,4-二氧六环(300ml)中,加入(e)-1,2-二溴乙烯
(sm-2,39.03g,0.21mol)的1,4-二氧六环(100ml)溶液,控温55~60℃反应,经检测反应完毕后,将反应液降温至室温,过滤,滤液加入纯化水(2000ml)中,二氯甲烷(600ml
×
3)提取,合并有机相,饱和食盐水(500ml
×
2)洗,无水硫酸钠干燥,过滤滤液减压浓缩至干即为中间体i-1,收率81.6%,纯度98.82%。
[0102]
化合物i-2的合成
[0103]
实施例9
[0104]
氩气保护室温条件下,将pd(pph3)2cl2(3.51g,5.0mmol)加入二甲基亚砜(200ml)中,搅拌混匀后,向反应溶剂中加入k3po4(59.43g,0.28mol)的纯化水(120ml)溶液,化合物i-1(37.83g,0.10mol)与2-(4-硼苯基)-2-甲基丙酸(sm-3,24.97g,0.12mol),控温90~95℃反应,经检测反应完毕后,过滤,将滤液加入纯化水(500ml)中,乙酸乙酯(200ml
×
3)提取,合并有机相,无水硫酸钠干燥,过滤,滤液减压浓缩至干后制得中间体i-2,收率96.6%,纯度99.85%。
[0105]
实施例10
[0106]
氩气保护室温条件下,将pd(pph3)4(5.78g,5.0mmol)加入二甲基亚砜(200ml)中,搅拌混匀后,向反应溶剂中加入k2co3(38.70g,0.28mol)的纯化水(50ml)溶液,化合物i-1(37.83g,0.10mol)与2-(4-硼苯基)-2-甲基丙酸(sm-3,20.02g,0.10mol),控温100~105℃反应,经检测反应完毕后,过滤,将滤液加入纯化水(500ml)中,甲基叔丁基醚(200ml
×
3)提取,合并有机相,无水硫酸钠干燥,过滤,滤液减压浓缩至干后制得中间体i-2,收率93.5%,纯度99.62%。
[0107]
实施例11
[0108]
氩气保护室温条件下,将pd(dppf)cl2(3.66g,5.0mmol)加入1,4-二氧六环(200ml)中,搅拌混匀后,向反应溶剂中加入na2co3(29.68g,0.28mol)的纯化水(130ml)溶液,化合物i-1(37.83g,0.10mol)与2-(4-硼苯基)-2-甲基丙酸(40.04g,0.2mol),控温85~90℃反应,经检测反应完毕后,过滤,将滤液加入纯化水(500ml)中,乙酸乙酯(200ml
×
3)提取,合并有机相,无水硫酸钠干燥,过滤,滤液减压浓缩至干后制得中间体i-2,收率92.9%,纯度99.56%。
[0109]
实施例12
[0110]
氩气保护室温条件下,将pd(dppf)cl2(3.66g,5.0mmol)加入二甲基亚砜(200ml)中,搅拌混匀后,向反应溶剂中加入naoac(22.96g,0.28mol)的纯化水(130ml)溶液,化合物i-1(37.83g,0.10mol)与2-(4-硼苯基)-2-甲基丙酸(44.04g,0.22mol),控温110~115℃反应,经检测反应完毕后,过滤,将滤液加入纯化水(500ml)中,氯仿(200ml
×
3)提取,合并有机相,无水硫酸钠干燥,过滤,滤液减压浓缩至干后制得中间体i-2,收率89.3%,纯度98.96%。
[0111]
实施例13
[0112]
氩气保护室温条件下,将pd(pph3)2cl2(3.51g,5.0mmol)加入二甲基亚砜(200ml)中,搅拌混匀后,向反应溶剂中加入k3po4(42.45g,0.20mol)的纯化水(100ml)溶液,化合物i-1(37.83g,0.10mol)与2-(4-硼苯基)-2-甲基丙酸(24.97g,0.12mol),控温95~100℃反应,经检测反应完毕后,过滤,将滤液加入纯化水(500ml)中,二氯甲烷(200ml
×
3)提取,合并有机相,无水硫酸钠干燥,过滤,滤液减压浓缩至干后制得中间体i-2,收率94.2%,纯度
99.61%。
[0113]
实施例14
[0114]
氩气保护室温条件下,将pd(pph3)2cl2(3.51g,5.0mmol)加入甲苯(200ml)中,搅拌混匀后,向反应溶剂中加入na3po4(57.38g,0.35mol)的纯化水(120ml)溶液,化合物i-1(37.83g,0.10mol)与2-(4-硼苯基)-2-甲基丙酸(24.97g,0.12mol),控温80~85℃反应,经检测反应完毕后,过滤,分液取有机相,纯化水(50ml
×
2)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干后制得中间体i-2,收率93.6%,纯度99.58%。
[0115]
实施例15
[0116]
氩气保护室温条件下,将pd(pph3)2cl2(3.51g,5.0mmol)加入甲苯(200ml)中,搅拌混匀后,向反应溶剂中加入na3po4(29.51g,0.18mol)的纯化水(120ml)溶液,化合物i-1(37.83g,0.10mol)与2-(4-硼苯基)-2-甲基丙酸(24.97g,0.12mol),控温75~80℃反应,经检测反应完毕后,过滤,分液取有机相,纯化水(50ml
×
2)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干后制得中间体i-2,收率90.6%,纯度99.88%。
[0117]
实施例16
[0118]
氩气保护室温条件下,将pd(pph3)2cl2(3.51g,5.0mmol)加入二甲基亚砜(200ml)中,搅拌混匀后,向反应溶剂中加入na3po4(60.66g,0.37mol)的纯化水(120ml)溶液,化合物i-1(37.83g,0.10mol)与2-(4-硼苯基)-2-甲基丙酸(24.97g,0.12mol),控温110~115℃反应,经检测反应完毕后,过滤,分液取有机相,纯化水(50ml
×
2)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干后制得中间体化合物i-2,收率89.9%,纯度98.88%。
[0119]
实施例17
[0120]
氩气保护室温条件下,将pd(pph3)2cl2(2.11g,3.0mmol)加入二甲基亚砜(200ml)中,搅拌混匀后,向反应溶剂中加入naoac(22.97g,0.28mol)的纯化水(80ml)溶液,化合物i-1(37.83g,0.10mol)与2-(4-硼苯基)-2-甲基丙酸(24.97g,0.12mol),控温90~95℃反应,经检测反应完毕后,过滤,将滤液加入纯化水(500ml)中,乙酸乙酯(200ml
×
3)提取,合并有机相,无水硫酸钠干燥,过滤,滤液减压浓缩至干后制得中间体i-2,收率93.8%,纯度99.72%。
[0121]
实施例18
[0122]
氮气保护室温条件下,将pd(pph3)2cl2(5.62g,8.0mmol)加入n,n-二甲基甲酰胺(200ml)中,搅拌混匀后,向反应溶剂中加入koac(27.48g,0.28mol)的纯化水(30ml)溶液,化合物i-1(37.83g,0.10mol)与2-(4-硼苯基)-2-甲基丙酸(24.97g,0.12mol),控温90~95℃反应,经检测反应完毕后,过滤,将滤液加入纯化水(500ml)中,乙酸乙酯(200ml
×
3)提取,合并有机相,无水硫酸钠干燥,过滤,滤液减压浓缩至干后制得中间体i-2,收率94.6%,纯度99.52%。
[0123]
实施例19
[0124]
氩气保护室温条件下,将pd(pph3)2cl2(0.70g,1.0mmol)加入二甲基亚砜(200ml)中,搅拌混匀后,向反应溶剂中加入naoac(22.97g,0.28mol)的纯化水(80ml)溶液,化合物i-1(37.83g,0.10mol)与2-(4-硼苯基)-2-甲基丙酸(sm-3,24.97g,0.12mol),控温75~80℃反应,经检测反应完毕后,过滤,将滤液加入纯化水(500ml)中,乙酸乙酯(200ml
×
3)提取,合并有机相,无水硫酸钠干燥,过滤,滤液减压浓缩至干后制得中间体i-2,收率89.6%,
纯度98.92%。
[0125]
实施例20
[0126]
氩气保护室温条件下,将pd(pph3)2cl2(7.01g,10.0mmol)加入二甲基亚砜(200ml)中,搅拌混匀后,向反应溶剂中加入koac(27.48g,0.28mol)的纯化水(80ml)溶液,化合物i-1(37.83g,0.10mol)与2-(4-硼苯基)-2-甲基丙酸(sm-3,24.97g,0.12mol),控温110~115℃反应,经检测反应完毕后,过滤,将滤液加入纯化水(500ml)中,乙酸乙酯(200ml
×
3)提取,合并有机相,无水硫酸钠干燥,过滤,滤液减压浓缩至干后制得中间体i-2,收率90.3%,纯度98.88%。
[0127]
化合物i的合成
[0128]
实施例21
[0129]
室温,将中间体i-2(23.08g,0.05mol)、环己烯(7.39g,0.09mol)、10%钯碳(2.31g)加入甲苯(250ml)中,控温105~110℃反应,经检测反应完毕后,将反应液降温至室温,过滤,滤液经纯化水(80ml
×
2)洗涤,饱和食盐水(80ml
×
2)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干即为目标产品比拉斯汀i,收率96.8%,纯度99.90%。
[0130]
实施例22
[0131]
室温,将中间体i-2(23.08g,0.05mol)、环己烯(5.75g,0.07mol)、10%钯碳(2.31g)加入二甲苯(250ml)中,控温125~130℃反应,经检测反应完毕后,将反应液降温至室温,过滤,滤液纯化水(80ml
×
2)洗涤,饱和食盐水(80ml
×
2)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干即为目标产品比拉斯汀i,收率92.3%,纯度99.63%。
[0132]
实施例23
[0133]
室温,将中间体i-2(23.08g,0.05mol)、环己烯(10.68g,0.13mol)、10%钯碳(2.31g)加入甲苯(250ml)中,控温105~110℃反应,经检测反应完毕后,将反应液降温至室温,过滤,滤液纯化水(80ml
×
2)洗涤,饱和食盐水(80ml
×
2)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干即为目标产品比拉斯汀i,收率93.0%,纯度99.60%。
[0134]
实施例24
[0135]
室温,将中间体i-2(23.08g,0.05mol)、环己烯(4.93g,0.06mol)、10%钯碳(2.31g)加入二甲苯(250ml)中,控温100~105℃反应,经检测反应完毕后,将反应液降温至室温,过滤,滤液纯化水(80ml
×
2)洗涤,饱和食盐水(80ml
×
2)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干即为目标产品比拉斯汀i,收率88.6%,纯度99.03%。
[0136]
实施例25
[0137]
室温,将中间体i-2(23.08g,0.05mol)、环己烯(11.50g,0.14mol)、10%钯碳(2.31g)加入甲苯(250ml)中,控温110~115℃反应,经检测反应完毕后,将反应液降温至室温,过滤,滤液纯化水(80ml
×
2)洗涤,饱和食盐水(80ml
×
2)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干即为目标产品比拉斯汀i,收率89.1%,纯度98.96%。
[0138]
实施例26
[0139]
室温,将中间体i-2(23.08g,0.05mol)、环己烯(7.39g,0.09mol)、10%钯碳(1.15g)加入甲苯(250ml)中,控温105~110℃反应,经检测反应完毕后,将反应液降温至室温,过滤,滤液纯化水(80ml
×
2)洗涤,饱和食盐水(80ml
×
2)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干即为目标产品比拉斯汀i,收率92.9%,纯度99.63%。
[0140]
实施例27
[0141]
室温,将中间体i-2(23.08g,0.05mol)、环己烯(7.39g,0.09mol)、10%钯碳(3.46g)加入甲苯(250ml)中,控温105~110℃反应,经检测反应完毕后,将反应液降温至室温,过滤,滤液纯化水(80ml
×
2)洗涤,饱和食盐水(80ml
×
2)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干即为目标产品比拉斯汀i,收率93.3%,纯度99.58%。
[0142]
实施例28
[0143]
室温,将中间体i-2(23.08g,0.05mol)、环己烯(7.39g,0.09mol)、10%钯碳(0.70g)加入甲苯(250ml)中,控温85~90℃反应,经检测反应完毕后,将反应液降温至室温,过滤,滤液纯化水(80ml
×
2)洗涤,饱和食盐水(80ml
×
2)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干即为目标产品比拉斯汀i,收率88.9%,纯度99.03%。
[0144]
实施例29
[0145]
室温,将中间体i-2(23.08g,0.05mol)、环己烯(7.39g,0.09mol)、10%钯碳(3.92g)加入甲苯(250ml)中,控温110~115℃反应,经检测反应完毕后,将反应液降温至室温,过滤,滤液经纯化水(80ml
×
2)洗涤,饱和食盐水(80ml
×
2)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干即为目标产品比拉斯汀i,收率87.4%,纯度98.92%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1