一种多官能度聚酯型紫外光固化树脂、组合物及其制备
本发明属于高分子材料技术领域,涉及一种多官能度聚酯型紫外光固化树脂、组合物及其制备。
背景技术:
近年来,有关紫外光固化技术的研究和开展日益活跃,在涂料、胶黏剂、微电子、齿科修复和生物材料等领域应用广泛。在来源方面,基本上所有丙烯酸酯都是石油基材料。如今,在环境污染和原油短缺的压力下,越来越关注对生物基材料的开发和应用,实现由再生物质向高分子材料和复合材料的转化。
高粘度是紫外光固化树脂的普遍问题。传统紫外固化树脂配方中的活性稀释剂比例很高,活性稀释剂的主要功能是降低系统的粘度,但是低分子量的活性稀释剂会导致高含量挥发性有机化合物排放,对环境造成影响。此外,活性稀释剂添加过多会降低固化后薄膜的柔韧性和机械性能。因此,亟需寻找方法来降低树脂粘度,减少活性稀释剂的使用。
cui等(polymerbulletin,2016,73,571-585)研究分析了分子链中引入了低分子量聚醚对树脂粘度的影响,发现低分子量聚醚的引入可以有效降低树脂粘度;long等(additivemanufacturing,2020,35)将胶体二氧化硅引入聚氨酯丙烯酸酯树脂基质中获得了一种聚合物-无机杂化材料,大大降低了体系的粘度。上述低分子量聚醚体系的uv树脂,虽然能有效降低树脂粘度,但受限于聚醚材料较低的内聚力,其固化材料的力学强度却普遍比较低。而无机胶体二氧化硅的引入,会降低树脂体系的相容性,同样会降低uv固化材料的力学性能。
技术实现要素:
本发明的目的就是为了提供一种多官能度聚酯型紫外光固化树脂、组合物及其制备,该紫外光固化树脂以生物基乳酸等为主体结构,将星形分子结构引入树脂中以降低粘度,其组合物作为光固化材料,除了具有固含量高、低气味、不含有机溶剂,可在紫外光辐射下快速固化的特点,还有较低的粘度,较好的涂层性能,是一种优质的绿色环保型树脂材料,可用于功能型涂料、胶黏剂等的研发和生产应用。
本发明所述的树脂,采用无规共聚的方式在分子链中引入聚乳酸和聚己内酯,通过聚己内酯组分降低树脂的整体玻璃化转变温度,并通过星形支化结构降低分子链之间的缠绕进一步降低树脂粘度;同时,本发明所述树脂由于采用共聚结构,可以有效避免相容性问题;进一步地,本发明所述树脂主体结构为内聚力更高的聚酯,因此固化后的力学性能极佳。
本发明的目的可以通过以下技术方案来实现:
本发明的技术方案之一提供了一种多官能度聚酯型紫外光固化树脂的制备方法,包括以下步骤:
(1)将聚酯单体加入到反应釜中,再加入多元醇与辛酸亚锡,在惰性气体保护下反应,得到聚酯多元醇;
(2)往聚酯多元醇中加入阻聚剂并分散,随后加入丙烯酸酐或甲基丙烯酸酐,继续在惰性气体下反应,反应结束后除去残余小分子,即得到目的产物。
进一步的,步骤(1)中,所述的聚酯单体为l-丙交酯、d-丙交酯、d,l-丙交酯和ε-己内酯中的一种或多种;
所述的多元醇为季戊四醇和双季戊四醇中的一种或两种。
进一步的,步骤(1)中,聚酯单体、多元醇和辛酸亚锡的添加量以重量份计,具体配比关系如下:
聚酯单体100份,
多元醇10-45份,
辛酸亚锡0.0001-0.001份。
进一步的,聚酯多元醇的数均分子量分子量范围为500~4000。
进一步的,步骤(1)中,反应温度为115℃,反应时间为6-12小时。
进一步的,步骤(2)中,所述的阻聚剂为对羟基苯甲醚、对苯二酚和2,6-二叔丁基-4-甲基苯酚中的一种或多种。
进一步的,步骤(2)中,聚酯多元醇、阻聚剂与丙烯酸酐或甲基丙烯酸酐以重量份计,具体配比关系如下:
聚酯多元醇100份
酸酐40-120份
阻聚剂0.2-0.5份
进一步的,步骤(2)中,反应温度为115℃,反应时间为3-5小时。
本发明的技术方案之二提供了一种多官能度聚酯型紫外光固化树脂,其采用如上所述的制备方法制备得到,其特征在于,该聚酯型紫外光固化树脂为含有多个丙烯酸酯官能团的聚酯型树脂。
本发明的技术方案之三提供了一种多官能度聚酯型紫外光固化树脂组合物,以重量份计,至少包括以下组分:
如上述的聚酯型紫外光固化树脂100份
自由基型紫外光引发剂3-5份
活性稀释剂10-30份
本发明的技术方案之四提供了一种多官能度聚酯型紫外光固化树脂组合物的制备方法,先取聚酯型紫外光固化树脂加入混合釜中,加热至40-60℃并保持10-20分钟,然后加入自由基型紫外光引发剂与活性稀释剂,搅拌均匀,真空除气泡,接着放入容器中封装,即得到目的产物。
进一步的,所述的自由基型紫外光引发剂为1-羟基-环已基-苯基甲酮、2-羟基-2-甲基-1-苯基丙酮、2-甲基-2-(4-吗啉基)-1-[4-(甲硫基)苯基]-1-丙酮和2,4,6-三甲基苯甲酰基-二苯基氧化膦中的一种或几种。
进一步的,所述的活性稀释剂为三羟甲基丙烷三丙烯酸酯、二缩三丙二醇二丙烯酸酯、二丙二醇二丙烯酸酯和1,6-己二醇二丙烯酸酯中的一种或几种。
聚酯单体在催化剂辛酸亚锡的作用下由多元醇引发剂引发聚合,其中聚酯单体对最终光固化树脂的力学性能有较大的影响。本发明中所用丙交酯属于生物发酵法生成的乳酸制备的生物基原料,所制备的聚乳酸树脂强度高、硬度大,所制备的聚己内酯树脂柔韧性好、相容性好,所制备的聚乳酸-己内酯共聚物在保持较高强度的同时,拥有较好的柔韧性;无规共聚的聚酯可以有效降低聚乳酸基树脂的粘度。多元醇作为引发剂,可引发聚酯单体无规共聚制备聚酯丙烯酸酯树脂,含多个羟基的多元醇能够为树脂引入星型结构,进一步降低树脂粘度。
反应工艺条件对整个反应过程也有非常大的影响,如反应温度过低,会导致反应时间的延长,从而导致生产周期变久,降低生产效率;而反应温度的提高,可能会导致副反应增加,甚至导致反应体系凝胶化。
与现有技术相比,本发明的树脂以生物基乳酸等为主体结构,将星形分子结构引入树脂中以降低粘度,其组合物作为光固化材料,除了具有固含量高、低气味、不含有机溶剂,可在紫外光辐射下快速固化的特点,还有较低的粘度,较好的涂层性能,是一种优质的绿色环保型树脂材料,可用于功能型涂料、胶黏剂等的研发和生产应用。
附图说明
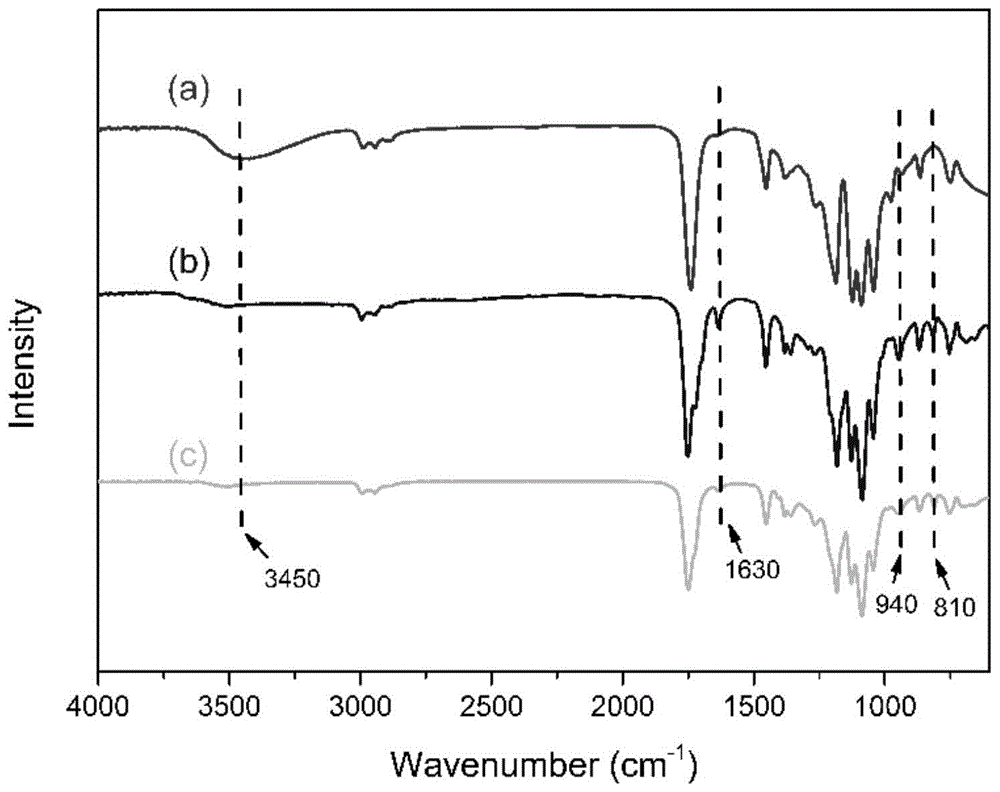
图1为实施例1制备的聚乳酸四元醇、四官能度聚乳酸甲基丙烯酸酯和紫外光固化后的树脂组合物的红外谱图;
图2为实施例1制备的聚乳酸四元醇和四官能度聚乳酸甲基丙烯酸酯的核磁谱图;
图3为实施例2制备的聚乳酸-己内酯四元醇、四官能度聚乳酸-己内酯甲基丙烯酸酯和紫外光固化后的树脂组合物的红外谱图
图4为实施例2制备的聚乳酸-己内酯四元醇和四官能度聚乳酸-己内酯甲基丙烯酸酯的核磁谱图。
具体实施方式
以下结合具体实施例对上述方案做进一步说明。应理解,这些实施例是用于说明本发明而不限于限制本发明的范围。实施例中采用的实施条件可以根据具体厂家的条件做进一步调整,未注明的实施条件通常为常规实验中的条件,此外,未具体说明的原料,也均为本领域的常规市售产品。
以下实施例所选的l-丙交酯、d-丙交酯和d,l-丙交酯由上海同杰良生物材料有限公司提供,所选的ε-己内酯、季戊四醇、双季戊四醇购自于阿达玛斯试剂有限公司,所选的辛酸亚锡购自于东京化成工业株式会社,所选的对羟基苯甲醚、对苯二酚和2,6-二叔丁基-4-甲基苯酚购自于上海泰坦科技股份有限公司,所选的丙烯酸酐和甲基丙烯酸酐购自于阿达玛斯试剂有限公司,所选的1-羟基-环已基-苯基甲酮、2-羟基-2-甲基-1-苯基丙酮、2-甲基-2-(4-吗啉基)-1-[4-(甲硫基)苯基]-1-丙酮和2,4,6-三甲基苯甲酰基-二苯基氧化膦购自于天津久日化学,所选的三羟甲基丙烷三丙烯酸酯、二缩三丙二醇二丙烯酸酯、二丙二醇二丙烯酸酯和1,6-己二醇二丙烯酸酯购自于长兴化学。
实施例1:
l-丙交酯100份
季戊四醇38份
辛酸亚锡0.001份
按上述重量配比称取l-丙交酯和季戊四醇,加入带有搅拌装置的反应釜中,再加入辛酸亚锡(0.15g),在氮气保护下于115℃反应12小时,得到聚乳酸四元醇。
聚乳酸四元醇100份
对苯二酚0.25份
甲基丙烯酸酐120份
再按上述重量配比向聚乳酸四元醇中加入对苯二酚,在搅拌下预分散10分钟,随后加入甲基丙烯酸酐,在氮气保护下于115℃反应4小时,反应结束后减压蒸馏去除残余的小分子,即得到聚乳酸甲基丙烯酸酯树脂.
再按上述重量配比将聚乳酸甲基丙烯酸酯树脂加入搅拌装置的反应釜中,加热至50℃,保持15分钟,待其粘度下降,加入2-羟基-2-甲基-1-苯基丙酮、三羟甲基丙烷三丙烯酸酯和二缩三丙二醇二丙烯酸酯,搅拌均匀后静置,除气泡,加入容器中进行封装,即得到聚乳酸甲基丙烯酸酯树脂组合物。
将聚乳酸甲基丙烯酸酯树脂组合物浇铸于固化模具中,放置于uv固化机中固化即可得到固化样品。经傅里叶红外光谱测试及nmr测试,成功制备聚乳酸甲基丙烯酸酯树脂,其组合物固化完全。
实施例2:
按上述重量配比取l-丙交酯,己内酯和季戊四醇,加入带有搅拌装置的反应釜中,再加入辛酸亚锡,在氮气保护下于115℃反应10小时,得到聚乳酸-己内酯四元醇;
聚乳酸-己内酯四元醇100份
对羟基苯甲醚0.4份
甲基丙烯酸酐70份
再按上述重量配比向聚乳酸-己内酯四元醇中加入对羟基苯甲醚,在搅拌下预分散10分钟,随后加入甲基丙烯酸酐,在氮气保护下于115℃反应4小时,反应结束后减压蒸馏去除残余的小分子,即得到聚乳酸-己内酯甲基丙烯酸酯树脂(pcla-ta)。
再按上述重量配比将聚乳酸-己内酯甲基丙烯酸酯树脂加入搅拌装置的反应釜中,加热至50℃,保持15分钟,待其粘度下降,加入2-羟基-2-甲基-1-苯基丙酮、三羟甲基丙烷三丙烯酸酯和二缩三丙二醇二丙烯酸酯,搅拌均匀后静置,除气泡,加入容器中进行封装,即得到聚乳酸-己内酯甲基丙烯酸酯树脂组合物。
将聚乳酸-己内酯甲基丙烯酸酯树脂组合物浇铸于固化模具中,放置于uv固化机中固化即可得到固化样品。经傅里叶红外光谱测试及nmr测试,成功制备聚乳酸-己内酯甲基丙烯酸酯树脂,其组合物固化完全。
实施例3:
己内酯100份
季戊四醇10份
辛酸亚锡0.001份
按上述重量配比取己内酯和季戊四醇,加入带有搅拌装置的反应釜中,再加入辛酸亚锡,在氮气保护下于115℃反应8小时,得到聚己内酯四元醇;
聚己内酯四元醇100份
2,6-二叔丁基-4-甲基苯酚0.4份
丙烯酸酐40份
再按上述重量配比向聚己内酯四元醇中加入2,6-二叔丁基-4-甲基苯酚,在搅拌下预分散10分钟,随后加入丙烯酸酐,在氮气保护下于115℃反应4小时,反应结束后减压蒸馏去除残余的小分子,即得到聚己内酯丙烯酸酯树脂。
再按上述重量配比将聚己内酯丙烯酸酯树脂加入搅拌装置的反应釜中,加热至50℃,保持15分钟,待其粘度下降,加入2-甲基-2-(4-吗啉基)-1-[4-(甲硫基)苯基]-1-丙酮、三羟甲基丙烷三丙烯酸酯和1二缩三丙二醇二丙烯酸酯,搅拌均匀后静置,除气泡,加入容器中进行封装,即得到聚己内酯丙烯酸酯树脂组合物。
将聚己内酯丙烯酸酯树脂组合物浇铸于固化模具中,放置于uv固化机中固化即可得到固化样品。经傅里叶红外光谱测试及nmr测试,成功制备聚己内酯丙烯酸酯树脂,其组合物固化完全。
实施例4:
d-丙交酯100份
双季戊四醇45份
辛酸亚锡0.001份
按上述重量配比取d-丙交酯和双季戊四醇,加入带有搅拌装置的反应釜中,再加入辛酸亚锡,在氮气保护下于115℃反应12小时,得到聚乳酸六元醇;
聚乳酸六元醇100份
2,6-二叔丁基-4-甲基苯酚0.5份
丙烯酸酐95份
按上述重量配比向聚乳酸六元醇中加入2,6-二叔丁基-4-甲基苯酚,在搅拌下预分散10分钟,随后加入丙烯酸酐,在氮气保护下于115℃反应5小时,反应结束后减压蒸馏去除残余的小分子,即得到聚乳酸丙烯酸酯树脂。
聚乳酸丙烯酸酯树脂100份
2,4,6-三甲基苯甲酰基-二苯基氧化膦3份
三羟甲基丙烷三丙烯酸酯20份
再按上述重量配比将聚乳酸丙烯酸酯树脂加入搅拌装置的反应釜中,加热至50℃,保持15分钟,待其粘度下降,加入2,4,6-三甲基苯甲酰基-二苯基氧化膦、三羟甲基丙烷三丙烯酸酯,搅拌均匀后静置,除气泡,加入容器中进行封装,即得到聚乳酸丙烯酸酯树脂组合物。
将聚乳酸丙烯酸酯树脂组合物浇铸于固化模具中,放置于uv固化机中固化即可得到固化样品。经傅里叶红外光谱测试及nmr测试,成功制备聚乳酸丙烯酸酯树脂,其组合物固化完全。
实施例5:
按上述重量配比取己内酯,l-丙交酯和双季戊四醇,加入带有搅拌装置的反应釜中,再加入辛酸亚锡,在氮气保护下于115℃反应11小时,得到聚乳酸-己内酯六元醇;
聚乳酸-己内酯六元醇100份
对羟基苯甲醚0.2份
甲基丙烯酸酐90份
按上述重量配比向聚乳酸-己内酯六元醇中加入对羟基苯甲醚,在搅拌下预分散10分钟,随后加入甲基丙烯酸酐,在氮气保护下于115℃反应4.5小时,反应结束后减压蒸馏去除残余的小分子,即得到聚乳酸-己内酯甲基丙烯酸酯树脂。
再按上述重量配比将聚乳酸-己内酯甲基丙烯酸酯树脂加入搅拌装置的反应釜中,加热至50℃,保持15分钟,待其粘度下降,加入聚乳酸-己内酯甲基丙烯酸酯树脂3wt%的1-羟基-环已基-苯基甲酮、10wt%的三羟甲基丙烷三丙烯酸酯和10wt%的二缩三丙二醇二丙烯酸酯,搅拌均匀后静置,除气泡,加入容器中进行封装,即得到聚乳酸-己内酯甲基丙烯酸酯树脂组合物。
将聚乳酸-己内酯甲基丙烯酸酯树脂组合物浇铸于固化模具中,放置于uv固化机中固化即可得到固化样品。经傅里叶红外光谱测试及nmr测试,成功制备聚乳酸-己内酯甲基丙烯酸酯树脂,其组合物固化完全。
实施例6:
按上述重量配比取己内酯,l-丙交酯和双季戊四醇,加入带有搅拌装置的反应釜中,再加入辛酸亚锡,在氮气保护下于115℃反应10小时,得到聚乳酸-己内酯六元醇;
聚乳酸-己内酯六元醇100份
对苯二酚0.45份
丙烯酸酐80份
按上述重量配比向聚乳酸-己内酯六元醇中加入对苯二酚,在搅拌下预分散10分钟,随后加入丙烯酸酐,在氮气保护下于115℃反应5小时,反应结束后减压蒸馏去除残余的小分子,即得到聚乳酸-己内酯丙烯酸酯树脂;
聚乳酸-己内酯丙烯酸酯树脂100份
2-甲基-2-(4-吗啉基)-1-[4-(甲硫基)苯基]-1-丙酮3份
三羟甲基丙烷三丙烯酸酯20份
按上述重量配比将聚乳酸-己内酯丙烯酸酯树脂加入搅拌装置的反应釜中,加热至50℃,保持15分钟,待其粘度下降,加入2-甲基-2-(4-吗啉基)-1-[4-(甲硫基)苯基]-1-丙酮、三羟甲基丙烷三丙烯酸酯,搅拌均匀后静置,除气泡,加入容器中进行封装,即得到聚乳酸-己内酯丙烯酸酯树脂组合物。
将聚乳酸-己内酯丙烯酸酯树脂组合物浇铸于固化模具中,放置于uv固化机中固化即可得到固化样品。经傅里叶红外光谱测试及nmr测试,成功制备聚乳酸-己内酯丙烯酸酯树脂,其组合物固化完全。
实施例7:
己内酯100份
季戊四醇20.5份
辛酸亚锡0.001份
按上述重量配比取己内酯和季戊四醇,加入带有搅拌装置的反应釜中,再加入辛酸亚锡,在氮气保护下于115℃反应7小时,得到聚己内酯四元醇;
聚乳酸四元醇100份
对羟基苯甲醚0.4份
甲基丙烯酸酐80份
按上述重量配比向聚己内酯四元醇中加入对羟基苯甲醚,在搅拌下预分散10分钟,随后加入一定量甲基丙烯酸酐,在氮气保护下于115℃反应4小时,反应结束后减压蒸馏去除残余的小分子,即得到聚己内酯甲基丙烯酸酯树脂。
再按上述重量配比将聚己内酯甲基丙烯酸酯树脂加入搅拌装置的反应釜中,加热至50℃,保持15分钟,待其粘度下降,加入2,4,6-三甲基苯甲酰基-二苯基氧化膦、三羟甲基丙烷三丙烯酸酯和二缩三丙二醇二丙烯酸酯,搅拌均匀后静置,除气泡,加入容器中进行封装,即得到聚己内酯甲基丙烯酸酯树脂组合物。
将聚己内酯甲基丙烯酸酯树脂组合物浇铸于固化模具中,放置于uv固化机中固化即可得到固化样品。经傅里叶红外光谱测试及nmr测试,成功制备聚己内酯甲基丙烯酸酯树脂,其组合物固化完全。
实施例8:
按上述重量配比取d,l-丙交酯,己内酯和季戊四醇,加入带有搅拌装置的反应釜中,再加入辛酸亚锡,在氮气保护下于115℃反应10小时,得到聚乳酸-己内酯四元醇;
聚乳酸-己内酯四元醇100份
对羟基苯甲醚0.4份
甲基丙烯酸酐70份
按上述重量配比向聚乳酸-己内酯四元醇中加入对羟基苯甲醚,在搅拌下预分散10分钟,随后加入甲基丙烯酸酐,在氮气保护下于115℃反应4小时,反应结束后减压蒸馏去除残余的小分子,即得到聚乳酸-己内酯甲基丙烯酸酯树脂(pcla-ta)。
再按上述重量配比将聚乳酸-己内酯甲基丙烯酸酯树脂加入搅拌装置的反应釜中,加热至50℃,保持15分钟,待其粘度下降,加入2-羟基-2-甲基-1-苯基丙酮、三羟甲基丙烷三丙烯酸酯和二缩三丙二醇二丙烯酸酯,搅拌均匀后静置,除气泡,加入容器中进行封装,即得到聚乳酸-己内酯甲基丙烯酸酯树脂组合物。
将聚乳酸-己内酯甲基丙烯酸酯树脂组合物浇铸于固化模具中,放置于uv固化机中固化即可得到固化样品。经傅里叶红外光谱测试及nmr测试,成功制备聚乳酸-己内酯甲基丙烯酸酯树脂,其组合物固化完全。
对比例1:
同实施例1,不同点在于,引发剂季戊四醇的用量为15.8重量份。
对比例2:
同实施例2,不同点在于,采用的引发剂是10重量份的1,4-丁二醇。
对比例3:
同实施例2,不同点在于,加入甲基丙烯酸酐后,反应温度为150℃。
对比例4:
同实施例2,不同点在于,将甲基丙烯酸酐改为等质量的马来酸酐。
对比例5:
同实施例2,不同点在于,省去了阻聚剂的加入。
图1为实施例1制备的聚乳酸四元醇、四官能度聚乳酸甲基丙烯酸酯和紫外光固化后的树脂组合物的红外谱图,其中,(a)聚乳酸四元醇、(b)四官能度聚乳酸甲基丙烯酸酯、(c)紫外光固化后的树脂组合物。如图所示,聚乳酸四元醇的红外谱图在3450cm-1处具有羟基强而宽的吸收峰,证实羟基封端预聚体被成功合成;在四官能度聚乳酸甲基丙烯酸酯的红外谱图中,1630cm-1处出现了不饱和c=c伸缩振动峰,810cm-1和940cm-1处出现了不饱和=ch2的振动峰,说明用甲基丙烯酸酐进行端基改性后,聚乳酸四元醇的端羟基已成功被丙烯酸酯基所取代。uv固化后,树脂的c=c双键和=c-h键的特征吸收峰减弱并消失,表明固化后,c=c在活性自由基的作用下发生自由基聚合,说明树脂的固化比较完全。;
图2为实施例1制备的聚乳酸四元醇和四官能度聚乳酸甲基丙烯酸酯的核磁谱图,其中,(a)为聚乳酸四元醇、(b)为四官能度聚乳酸甲基丙烯酸酯。在聚乳酸四元醇的核磁谱图中,化学位移5.17ppm处为聚乳酸链段中次甲基-ch-的质子峰,1.59ppm处为聚乳酸链段中甲基ch3的质子峰;证实该预聚体的主链结构是聚乳酸;而4.37ppm和1.50ppm处较弱的两个峰,则是由邻近端羟基处聚乳酸单元中的质子引起的,符合羟基封端的聚乳酸四元醇的结构特征。在聚乳酸甲基丙烯酸酯的核磁谱图中,在5.68ppm和6.24ppm处出现了新的特征峰,对应于甲基丙烯酸酯基团上直接与c=c双键相连的亚甲基ch2中两个质子;在1.96处出现了新的特征峰,对应于甲基丙烯酸酯基团上直接与c=c双键相连的甲基ch3中的质子,证实聚乳酸四元醇的端羟基被甲基丙烯酸酯基团成功取代。;
图3为实施例2制备的聚乳酸-己内酯四元醇、四官能度聚乳酸-己内酯甲基丙烯酸酯和紫外光固化后的树脂组合物的红外谱图,其中,(a)为聚乳酸-己内酯四元醇、(b)为四官能度聚乳酸-己内酯甲基丙烯酸酯、(c)为紫外光固化后的树脂组合物。聚乳酸-己内酯四元醇的红外谱图在3400-3500cm-1附近的宽吸收峰可归因于末端-oh的伸缩振动,表明聚乳酸-己内酯四元醇成功合成;与图1类似,聚乳酸-己内酯甲基丙烯酸酯的红外谱图在816cm-1和940cm-1处出现了不饱和=ch2弯曲振动和伸缩振动吸收峰,证明聚乳酸-己内酯二元醇的端羟基成功被丙烯酸酯取代;uv光固化后,这些双键特征峰又消失,说明该树脂发生了固化反应。
图4为实施例2制备的聚乳酸-己内酯四元醇和四官能度聚乳酸-己内酯甲基丙烯酸酯的核磁谱图,(a)为聚乳酸-己内酯四元醇,(b)为四官能度聚乳酸-己内酯甲基丙烯酸酯。聚乳酸-己内酯四元醇的核磁谱图中,化学位移5.04ppm处为聚乳酸链段中次甲基-ch-的质子峰,1.46ppm处为聚乳酸链段中甲基ch3的质子峰;化学位移1.43ppm、1.66ppm、2.28ppm和4.15ppm均为聚己内酯链段中亚甲基ch2的质子峰,说明该预聚物是由聚乳酸和聚己内酯组成的共聚物;此外,在化学位移3.58ppm和4.21ppm处分别出现了两处较小的特征峰,分别对应于聚己内酯链端基上邻近羟基处亚甲基ch2中质子峰和聚乳酸链端基上邻近羟基处次甲基ch中质子峰,说明该预聚物分子链中聚乳酸和聚己内酯链段是无规排列在端基处的,属于无规共聚物,符合羟基封端的聚乳酸-己内酯四元醇的结构特征。在聚乳酸-己内酯甲基丙烯酸酯的核磁谱图中,在6.17ppm,5.60ppm和1.86ppm处出现了新的特征峰,分别对应于甲基丙烯酸酯基团上直接与c=c双键相连的亚甲基ch2和甲基ch3中的质子峰,证实聚乳酸-己内酯四元醇的端羟基被甲基丙烯酸酯基团成功取代。
性能测试
分子量:采用agilent1260型液相色谱仪测定聚酯丙烯酸酯树脂的分子量。
粘度:采用brookfield公司的dv-i型旋转粘度计测定聚酯丙烯酸酯树脂在25℃温度下的粘度。
形态:肉眼观察25℃下聚酯丙烯酸酯树脂的物理形态。
拉伸强度:根据gb/t1040.3-2006,采用wwd3020型万能拉力机对uv固化后的树脂样条进行拉伸测试。
涂层附着力:根据iso2409:1992国际标准,采用百格法对uv固化后的涂层进行测试。
涂层铅笔硬度:根据gb/t6739-2006,用铅笔硬度仪测定uv固化后的漆膜硬度。
涂层柔韧性:根据gb/t1731-1993,测定uv固化后涂层的柔韧性。
表1实施例1-8的多官能度聚酯型紫外光固化树脂及其组合物的性能测试表
由表1可以看出,本发明中的多官能度聚酯型紫外光固化树脂及其组合物在具有低粘度的同时,所得到的uv固化制品也具有优异的力学强度、涂层附着力和良好的涂层韧性与硬度。
从对比例1可以看出,当聚酯基树脂的分子量增加时,树脂粘度会急剧增大,特别是纯聚乳酸基聚酯,粘度增加更加明显,过高的粘度不利于施工,会使组合物固化不完全,从而造成力学强度大幅降低,涂层附着力也会下降。
从对比例2可以看出,在树脂分子量与对比例1相近时,共聚可以有效降低树脂粘度;但对比例2与实施例2相比,将四官能度的引发剂改为了二官能度,仍然导致了树脂粘度的急剧增加,从而不利于施工,组合物的固化不良,从而造成力学强度大幅降低,涂层附着力也会下降,涂层硬度也比较低。
从对比例3可以看出,反应温度的升高,导致反应凝胶现象的出现,无法有效制备得到星型光固化树脂,无法测试性能。
从对比例4可以看出,引入等质量的马来酸酐会使粘度有少量上升,且无法在紫外光辐射下有效固化,无法测试性能。
从对比例5可以看出,缺少阻聚剂的加入,会使树脂的储存稳定性变差,在合成过程中或储运过程中提前发生聚合反应,从而导致凝胶化,无法使用。
以上各实施例中,各原料配方还可以根据需要在以下配比范围内任意调整,即可以任意调整为对应范围的端值或中间点值:
前面的实例仅是说明性的,用于解释本发明所述方法的一些特征。所附的权利要求旨在要求尽可能广的范围,且本文所呈现的实施例仅是根据所有可能的实施例组合的选择的实施方式的说明。因此,申请人的用意是所附的权利要求不被说明本发明的特征的示例的选择限制。在权利要求中所用的一些数值范围也包括了在其范围之内的子范围,这些范围中的变化也应在可能的状况下解释为被所附的权利要求覆盖。
- 还没有人留言评论。精彩留言会获得点赞!