一种肽核酸鸟嘌呤单体的合成方法与流程
1.本发明属于化学合成技术领域,尤其是涉及一种肽核酸鸟嘌呤单体的合成方法。
背景技术:
2.肽核酸,一类以多肽骨架取代糖磷酸主链的dna类似物, 是丹麦有机化学家 ole buchardt和生物化学家 peter nielsen 于20 世纪80 年代开始潜心研究的一种新的核酸序列特异性试剂。它是在第一代、第二代反义试剂的基础上 ,通过计算机设计构建并最终人工合成的第三代反义试剂,是一种全新的dna类似物,即以中性的肽链酰胺2-氨基乙基甘氨酸键取代了dna中的戊糖磷酸二酯键骨架,其余的与dna相同,pna可以通过watson-crick碱基配对的形式识别并结合dna或rna序列,形成稳定的双螺旋结构。
3.根据pna的代谢稳定性,主要将其用于抑制基因表达的反义药物研究领域,国外几家制药及生物技术公司均投入大量精力从事开发研究;根据其与dna优良的杂交稳定性,pna又被广泛用于dna分子识别和操纵。pna可以广泛用于病原体、遗传病检测的分子杂交、原位杂交、突变分析、抗癌、抗病毒反义核酸研究和应用。尤其是pna可以取代寡核苷酸用于基因芯片的制备,将比普通基因芯片更稳定,特异性也更好,被认为是基因芯片的升级产品。pna也可用于定量pcr,可以用于实时检测pcr的扩增反应;也可将其做成pna beacon,用于实时监测细胞内的rna表达。随着pna基础研究的不断深入和新技术的不断出现,pna将显示出无比优越的性能和更为广阔的应用前景。
4.由于 pna 的特殊化学结构且不带电荷,所以 pna 探针与目标 dna 结合的特异性与灵敏性都非常高。因此pna 作为 fish probes 非常有效,即使在浓度非常低的情况下也能够有比较明显的效果。pna probes 与目标序列的结合非常快 (几个小时内),而且干扰背景少。
5.目前世界范围内新冠肺炎蔓延,肽核酸探针作为快速诊断试剂必将得到广泛应用。
6.目前国内没有一家公司能对这个产品进行大规模的产业化,其合成规模一般只达到克级水平,仅适合于研究用,无法提供长期稳定的商品供应。其中合成工艺路线长,操作繁琐,是大规模合成肽核酸鸟嘌呤单体的瓶颈。
7.现有肽核酸鸟嘌呤单体合成工艺存在着诸多问题,合成路线长,工艺复杂,收率低,操作繁琐;反应中用到无水无氧条件;投料比例难以控制,造成副产物增多。尤其是合成最终产物的步骤,由于两个保护基在反应条件下都有可能脱落,使得反应收率难以把握,其后处理中杂质难以去除。
技术实现要素:
8.有鉴于此,本发明旨在提出一种肽核酸鸟嘌呤单体的合成方法,以克服现有技术中的不足,避免了无水无氧反应条件的使用,反应条件温和;溶剂可以重复再利用,节约生产成本;工艺路线得到简化;为肽核酸鸟嘌呤单体的合成的产业化提供基础。
9.为达到上述目的,本发明的技术方案是这样实现的:一种肽核酸鸟嘌呤单体的合成方法,包括如下步骤,1)将2-n-(二苯甲氧羰基)鸟嘌呤-9-乙酸溶于n,n-二甲基甲酰胺中,搅拌下加入n-羟基丁二酰亚胺,0℃下加入1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐,在室温下搅拌反应,tlc显示反应完毕;2)将n-(2-fmoc-氨乙基)甘氨酸溶于n,n-二甲基甲酰胺中,加入步骤1)得到的活性酯,0℃下滴加n,n-二异丙基乙胺,室温反应过夜,即得到所需的产物肽核酸鸟嘌呤单体。
10.优选的,步骤1)中,所述2-n-(二苯甲氧羰基)鸟嘌呤-9-乙酸与n-羟基丁二酰亚胺的摩尔比为1:1。
11.优选的,步骤1)中,所述2-n-(二苯甲氧羰基)鸟嘌呤-9-乙酸与1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐的摩尔比为1:3。
12.优选的,步骤2)中,所述n-(2-fmoc-氨乙基)甘氨酸与活性酯的摩尔比为1:1。
13.优选的,步骤2)中,所述n-(2-fmoc-氨乙基)甘氨酸与n,n-二异丙基乙胺的摩尔比为1:1。
14.合成工艺中的反应式如下所示,相对于现有技术,本发明所述的一种肽核酸鸟嘌呤单体的合成方法,具有以下优势:1、本发明简化了原有的合成工艺路线,避免了繁琐的的操作步骤。
15.2、本发明减少了反应原料种类,且原料易得,价格低廉。溶剂可以重复利用,实现了绿色合成的目的。
16.3、本发明避免了合成工艺中无水无氧反应条件的使用,反应条件温和。
17.4、本发明后处理简单便捷,能得到高品质的产物,且此方法可以放大操作,实现工业化。
附图说明
18.构成本发明的一部分的附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
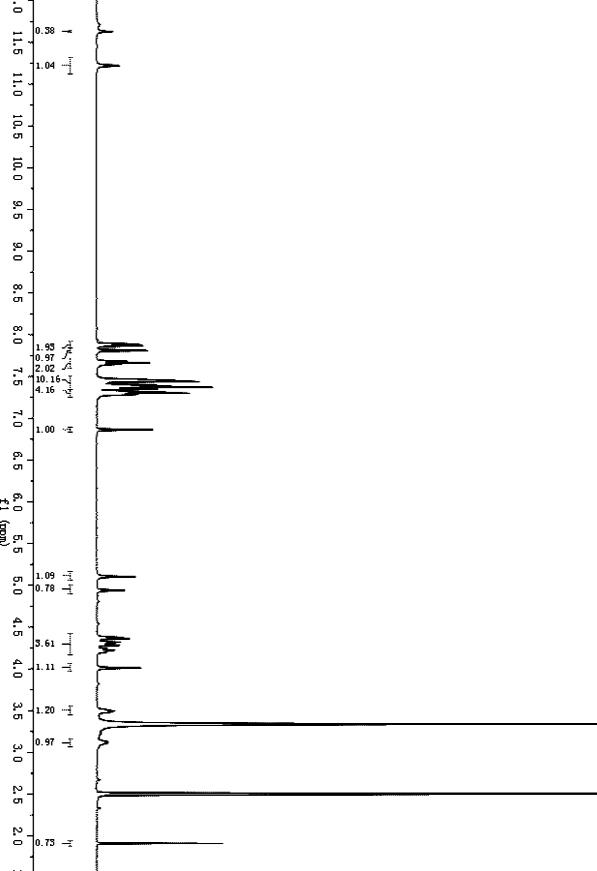
19.在附图中:图1为本发明实施例一所述的肽核酸鸟嘌呤单体的核磁数据图。
具体实施方式
20.需要说明的是,在不冲突的情况下,本发明中的实施例及实施例中的特征可以相互组合。
21.下面将参考附图并结合实施例来详细说明本发明。
22.实施例一一种肽核酸鸟嘌呤单体的合成方法,包括如下步骤:1)将41.9克2-n-(二苯甲氧羰基)鸟嘌呤-9-乙酸溶于200毫升n,n-二甲基甲酰胺中,搅拌下加入11.5克n-羟基丁二酰亚胺,0℃下加入57.6克1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐,在室温下搅拌反应,tlc显示反应完毕,将析出的固体过滤,水洗一次,得到2-n-(二苯甲氧羰基)鸟嘌呤-9-乙酸nhs活性酯,无需进一步纯化,直接用于下一步反应,母液减压蒸馏回收溶剂;2)将34克n-(2-fmoc-氨乙基)甘氨酸溶于200毫升n,n-二甲基甲酰胺中,加入步骤1)得到的nhs活性酯,0℃下滴加12.9克n,n-二异丙基乙胺,缓慢升至室温,室温反应过夜,将溶剂减压下除去,除去的溶剂可以回收再利用,将得到粗产物水中打浆,过滤,水洗三次,烘干得到所需的产物肽核酸鸟嘌呤单体33.2克,收率44.6%。
23.由核磁谱图,如图1可以看出,产物氢谱特征峰很明显:一个羧酸活泼氢和一个杂环氮氢活泼氢出现在化学位移11-12的位置;芳香区共有20个氢,为保护基苯环以及两个酰胺键的氮氢;脂肪链区共有8个亚甲基峰比较清晰。
24.根据核磁分析可知,通如上步骤得到了高纯度的目标产物。实施例2一种肽核酸鸟嘌呤单体的合成方法,包括如下步骤:1)将41.9克2-n-(二苯甲氧羰基)鸟嘌呤-9-乙酸溶于150毫升n,n-二甲基甲酰胺中,搅拌下加入12.5克n-羟基丁二酰亚胺,0℃下加入57.6克1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐,在室温下搅拌反应,tlc显示反应完毕,将析出的固体过滤,水洗一次,得到2-n-(二苯甲氧羰基)鸟嘌呤-9-乙酸nhs活性酯,无需进一步纯化,直接用于下一步反应,母液减压蒸馏回收溶剂;2)将34克n-(2-fmoc-氨乙基)甘氨酸溶于150毫升n,n-二甲基甲酰胺中,加入步骤1)得到的nhs活性酯,0℃下滴加12.9克n,n-二异丙基乙胺,缓慢升至室温,室温反应过夜,将溶剂减压下除去,除去的溶剂可以回收再利用,将得到粗产物水中打浆,过滤,水洗三次,烘干得到所需的产物肽核酸鸟嘌呤单体32.8克,收率44.3%。
25.实施例3一种肽核酸鸟嘌呤单体的合成方法,包括如下步骤:1)将41.9克2-n-(二苯甲氧羰基)鸟嘌呤-9-乙酸溶于150毫升n,n-二甲基甲酰胺中,搅拌下加入11.5克n-羟基丁二酰亚胺,0℃下加入60.6克1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐,在室温下搅拌反应,tlc显示反应完毕,将析出的固体过滤,水洗一次,得到2-n-(二苯甲氧羰基)鸟嘌呤-9-乙酸nhs活性酯,无需进一步纯化,直接用于下一步反应,母液减压蒸馏回收溶剂;2)将34克n-(2-fmoc-氨乙基)甘氨酸溶于150毫升n,n-二甲基甲酰胺中,加入步骤1)得到的nhs活性酯,0℃下滴加12.9克n,n-二异丙基乙胺,缓慢升至室温,室温反应过夜,将溶剂减压下除去,除去的溶剂可以回收再利用,将得到粗产物水中打浆,过滤,水洗三次,烘干得到所需的产物肽核酸鸟嘌呤单体33.2克,收率44.8%。
26.实施例4一种肽核酸鸟嘌呤单体的合成方法,包括如下步骤:
1)将41.9克2-n-(二苯甲氧羰基)鸟嘌呤-9-乙酸溶于150毫升n,n-二甲基甲酰胺中,搅拌下加入11.5克n-羟基丁二酰亚胺,0℃下加入57.6克1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐,在室温下搅拌反应,tlc显示反应完毕,将析出的固体过滤,水洗一次,得到2-n-(二苯甲氧羰基)鸟嘌呤-9-乙酸nhs活性酯,无需进一步纯化,直接用于下一步反应,母液减压蒸馏回收溶剂;2)将37克n-(2-fmoc-氨乙基)甘氨酸溶于150毫升n,n-二甲基甲酰胺中,加入步骤1)得到的nhs活性酯,0℃下滴加12.9克n,n-二异丙基乙胺,缓慢升至室温,室温反应过夜,将溶剂减压下除去,除去的溶剂可以回收再利用,将得到粗产物水中打浆,过滤,水洗三次,烘干得到所需的产物肽核酸鸟嘌呤单体31.8克,收率42.7%。
27.实施例五一种肽核酸鸟嘌呤单体的合成方法,包括如下步骤:1)将41.9克2-n-(二苯甲氧羰基)鸟嘌呤-9-乙酸溶于150毫升n,n-二甲基甲酰胺中,搅拌下加入11.5克n-羟基丁二酰亚胺,0℃下加入57.6克1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐,在室温下搅拌反应,tlc显示反应完毕,将析出的固体过滤,水洗一次,得到2-n-(二苯甲氧羰基)鸟嘌呤-9-乙酸nhs活性酯,无需进一步纯化,直接用于下一步反应,母液减压蒸馏回收溶剂;2)将34克n-(2-fmoc-氨乙基)甘氨酸溶于150毫升n,n-二甲基甲酰胺中,加入步骤1)得到的nhs活性酯,0℃下滴加14.9克n,n-二异丙基乙胺,缓慢升至室温,室温反应过夜,将溶剂减压下除去,除去的溶剂可以回收再利用,将得到粗产物水中打浆,过滤,水洗三次,烘干得到所需的产物肽核酸鸟嘌呤单体31.7克,收率43.2%。
28.实施例2~实施例5得到的产物的表征结果与实施例1一致。
29.以上所述仅为本发明创造的较佳实施例而已,并不用以限制本发明创造,凡在本发明创造的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明创造的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1