一种粘弹性荧光共轭聚合物及其制备方法
本发明涉及共轭聚合物制备领域,尤其涉及一种粘弹性荧光共轭聚合物及其制备方法。
背景技术:
共轭聚合物是一类有机半导体,是一类不饱和的聚合物,在其主链上所有的原子都是sp-或sp2-杂化,在其本征态、中性状态都是绝缘体或者是宽带隙半导体,只有在掺杂后才能成为导体。因此共轭聚合物在光学、电子学、光电、光子器件、传感等领域得到广泛应用,比如发光二极管、薄膜晶体管、太阳能电池等领域。
共轭聚合物具有独特的光电性能,其本征上有刚硬性的共轭骨架构成,但是现有的共轭聚合物通常是粉末状态,因此其延展性较差,限制了共轭聚合物在柔性器件上的应用。
因此,现有技术还有待于改进和发展。
技术实现要素:
鉴于上述现有技术的不足,本发明的目的在于提供一种粘弹性荧光共轭聚合物及其制备方法,旨在提高现有的共轭聚合物的延展性以适应当前对柔性材料的需求。
本发明的技术方案如下:
一种粘弹性荧光共轭聚合物,其中,所述粘弹性荧光共轭聚合物的化学结构式为:
一种如上所述的粘弹性荧光共轭聚合物的制备方法,其中,包括步骤:
a、在惰性气氛下,将单体:
b、在惰性气氛下,将1,3-双(二苯基膦基)丙烷氯化镍加入到格氏反应后的溶液中进行催化转移缩聚反应,得到第二混合体系;
c、将催化转移缩聚反应后的溶液进行纯化处理,得到所述共轭聚合物
所述的制备方法,其中,步骤a中,所述格氏反应的温度为0℃,时间为2~3h。
所述的制备方法,其中,步骤a中,所述单体与所述格氏试剂的摩尔比为1:1~1.5
所述的制备方法,其中,步骤b中,所述催化转移缩聚反应的温度为40-60℃,时间为10-14h。
所述的制备方法,其中,步骤b中,所述单体与所述1,3-双(二苯基膦基)丙烷氯化镍的摩尔比为50~700:1。
所述的制备方法,其中,步骤a中,所述有机溶剂为四氢呋喃、甲苯或氯苯中的一种。
所述的制备方法,其中,步骤c中,所述纯化处理的步骤包括:
c1、将催化转移缩聚反应后的溶液加入到丙酮中并进行离心处理,去除上清液后进行干燥,得到聚合物粗产物;
c2、将所述聚合物粗产物溶于三氯甲烷,采用制备型高效液相色谱对聚合物粗产物进行提纯后再进行干燥。
所述的制备方法,其中,步骤c1中,所述离心的速度为5500-6500r/min,离心的时间为10-15min。
一种如上所述的粘弹性荧光共轭聚合物的应用,其中,用于制备柔性器件或柔性可穿戴设备。
有益效果:本发明制备的粘弹性荧光共轭聚合物以噻吩基团作为共轭骨架,噻吩基团的侧链上具有两条较长的碳支链,形成庞大的侧链基团,使共轭聚合物在室温无溶剂状态下展现出较好的延展性,同时由于庞大烷基侧链的存在,聚合物骨架展现出区域规整性,并且可根据聚合物分子量的大小来实现材料柔性程度的调节。本发明的粘弹性共轭聚合物的制备方法简单易实现,且能耗较小,可应用于柔性发光器件或柔性可穿戴设备中。
附图说明
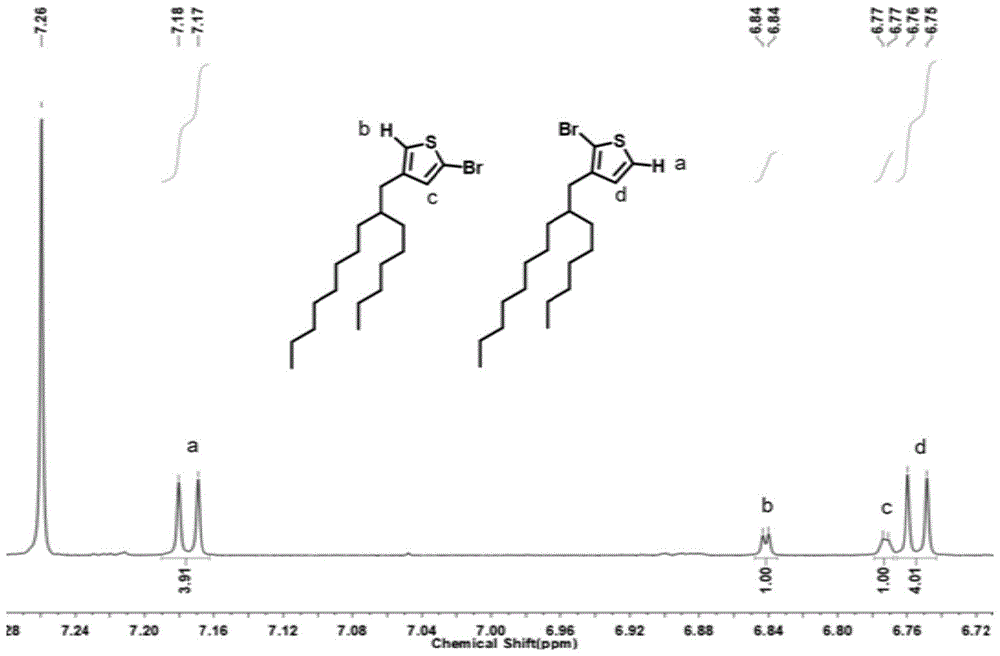
图1为本发明的粘弹性荧光共轭聚合物的格氏反应后的中间体的核磁光谱图。
图2为本发明的不同分子量的粘弹性荧光共轭聚合物的核磁光谱图。
图3为本发明的不同分子量的粘弹性荧光共轭聚合物的凝胶渗透色谱图曲线。
图4(a,b)为本发明的不同分子量的粘弹性荧光共轭聚合物的dsc保留时间曲线图;其中,(a)为冷却过程的保留时间,(b)为加热过程的保留时间。
图5(a,b)为本发明的粘弹性荧光共轭聚合物分子量(mn,gpc)与玻璃化转变温度(tg)之间关系的方程模型关系图;其中,(a)为flory-fox和fox-loshaek方程模型关系图,(b)为ogawa方程模型关系图。
具体实施方式
本发明提供一种粘弹性荧光共轭聚合物及其制备方法,为使本发明的目的、技术方案及效果更加清楚、明确,以下对本发明进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
本实施例提供一种粘弹性荧光共轭聚合物,所述粘弹性荧光共轭聚合物的化学结构式为:
具体说来,现有的共轭聚合物由于存在共轭基团,链刚性大,结晶性强,难以满足对柔性共轭聚合物材料的需求,严重阻碍了柔性共轭聚合物材料的商业化发展。本实施例的粘弹性荧光共轭聚合物中,以噻吩基团作为共轭骨架,噻吩基团的侧链上具有两条较长的碳支链,形成庞大的侧链基团。碳支链上的碳原子数限定为x1=x2+2,x2的取值为3、5、7、11,换句话说,叔碳上的两条烷烃链中,其中一条烷烃链上的碳原子数是4、6、8、12,另一条烷烃链上的碳原子数比前一条烷烃链多2个碳原子。这些侧链基团作为“增塑剂”,使共轭聚合物在室温下具有粘弹性,且在室温无溶剂状态下展现出了优异的延展性,其玻璃化转变温度变化规律与普通粘弹性聚合物的玻璃化转变温度的变化规律相同,可应用于柔性发光器件或柔性可穿戴设备中。同时由于庞大烷基侧链的位阻效应,使得聚合物的骨架排列上具有区域规整性的特点。本发明的共轭聚合物还易溶于常用的有机溶剂中,如四氢呋喃、二氯甲烷、三氯甲烷、氯苯,因此具有较好的溶液可加工性。
本发明实施例提供一种如上所述的粘弹性荧光共轭聚合物的制备方法,其中,包括步骤:
s10、在惰性气氛下,将单体
s20、在惰性气氛下,再将催化剂1,3-双(二苯基膦基)丙烷氯化镍加入到格氏反应后的溶液中进行催化转移缩聚反应;
s30、将催化转移缩聚反应后的溶液进行纯化处理,得到所述共轭聚合物
本发明是基于噻吩基团带有庞大烷基侧链的单体原料,并通过催化转移缩聚反应,制备出一种可控分子量和区域规整性的粘弹性荧光共轭聚合物。所述催化转移缩聚反应的反应温度低,反应时间短,且通过改变单体和催化剂的用量比例可调整共轭聚合物聚合物分子量的大小,以实现对共轭聚合物的柔性程度的调节,从而适应不同领域对柔性共轭聚合物的需求。同时,由于噻吩基团上的烷基侧链基团具有较大的位阻效应,在催化转移缩聚反应过程中保证了合物共轭骨架的区域规整性,提高了共轭聚合物的荧光特性。并且本发明提供的粘弹性荧光共轭聚合物的制备方法简单易实现,能耗较小。
在一种实施方式中,步骤s10中,所述格氏反应的温度为0℃,时间为2~3h。
在一种实施方式中,步骤s10中,所述单体与所述格氏试剂的摩尔比为1:1~1.5
在一种实施方式中,步骤s20中,所述催化转移缩聚反应的温度为40~60℃,时间为10~14h。当温度高于60℃时,反应速度过快不易控制,当温度低于40℃时,则催化剂活性较低,反应效率较低。优选的,催化转移缩聚反应的温度为50℃,反应时间为12h,在该条件下,催化转移缩聚反应既可控又高效。
在一种实施方式中,步骤s20中,所述单体与所述催化剂的摩尔比为50~800:1。优选的,为节约成本并保证实现不同分子量的可控聚合,在进行催化转移缩聚反应时,所述单体和催化剂的摩尔比为50:1、100:1、125:1、150:1、300:1或700:1。
在一种实施方式中,步骤s10和s20中,所述惰性气氛为氮气、氩气、氖气和氦气中的一种或多种。
在一种实施方式中,步骤s10中,所述有机溶剂包括但不限于四氢呋喃(thf)、甲苯或氯苯中的一种。
在一种实施方式中,步骤s30中,所述纯化处理的步骤包括:
s31、将催化转移缩聚反应后的溶液加入到丙酮中并进行离心处理,去除上清液后进行干燥,得到聚合物粗产物;
s32、将所述聚合物粗产物溶于三氯甲烷,采用制备型高效液相色谱对聚合物粗产物进行提纯后再进行干燥。
具体的,将催化转移缩聚反应后的溶液逐滴滴入盛有丙酮的离心管中,使共轭聚合物析出,离心后将上层清液倒掉后,获得聚合物粗产物,将聚合物粗产物放入真空干燥箱进行真空干燥;将干燥后的聚合物粗产物用溶解于少量三氯甲烷中制成溶液,通过玻璃注射器将所述溶液注入制备型高效液相色谱(phplc)中进行提纯,以除去聚合物粗产物中的少量低聚物和未反应的单体,得到提纯后的共轭聚合物,再将提纯后的共轭聚合物放入真空干燥箱进行真空干燥。优选的,所述离心的速度为5500-6500r/min,离心的时间为10-15min。
本发明还提供一种如上所述的粘弹性荧光共轭聚合物的应用,所述粘弹性荧光共轭聚合物可应用于制备柔性器件或柔性可穿戴设备。
下面通过具体实施例对本发明进行详细说明。
实施例1
聚(3-(2-己基癸基)噻吩)(pt-c6c10)的合成,反应式为:
(1)在氩气气氛下,向烧瓶中装入单体2,5-二溴-3-(2-己基癸基)噻吩(400mg,0.86mmol)。通过注射器向烧瓶中加入无水thf(5.0ml),并将混合物在0℃下搅拌均匀。再通过注射器向该混合物体系中加入异丙基氯化镁(异丙基氯化镁溶液为浓度为2.0m的thf溶液;投量为51ml,1.02mmol),并在0℃下搅拌30分钟,然后在室温下搅拌2小时。
(2)将1,3-双(二苯基膦基)丙烷氯化镍(ii)(ni(dppp)cl2,9.3mg,0.017mmol,2mol%)加入到无水thf(3.0ml)中,制成ni(dppp)cl2的thf悬浮液,并通过注射器将所述悬浮液加入到格氏反应后的溶液中。在室温下,溶液颜色立即从浅黄色变为红色。再将反应温度升高至50℃,搅拌12小时。
(3)将催化转移缩聚反应后的溶液冷却至室温后,用5m盐酸淬灭反应加入5m盐酸,并用chcl3进行萃取,采用去离子水洗涤有机层,萃取后将分液漏斗中的有机层收集,并用无水mgso4粉末对收集到的有机层溶液进行干燥,过滤并在减压条件下浓缩,将浓缩后的溶液加入到丙酮中,通过多次离心处理(6000rpm,15分钟)并除去上清液,得到聚合物粗产物,将聚合物粗产物放入40℃的真空干燥箱中进行真空干燥12h。然后,将干燥后的聚合物粗产物溶于少量三氯甲烷中制成溶液,并通过玻璃注射器将所述溶液注入制备型高效液相色谱(phplc)进行提纯,得到提纯后的粘弹性荧光共轭聚合物(棕红色液体),产率约为10%,记为p1。该粘弹性荧光共轭聚合物在室温条件下为深橙色的焦油状液体,在365nm紫外线下显示出桔黄色至橙色的荧光。
实施例2
不同分子量的聚(3-(2-己基癸基)噻吩)(pt-c6c10)的合成。
具体合成步骤如实施例1所述,其中,通过改变单体与催化剂的投料比,来实现对不同分子量的pt-c6c10可控聚合,并将不同分子量的pt-c6c10记为p1~p6,具体条件见表1所示,其中,p1为实施例1制备的pt-c6c10。
表1不同反应条件的聚合反应
将实施例1和实施例2中制备的具有不同分子量的pt-c6c10进行表征,结果如下:
(1)采用核磁光谱仪(jeolecs-400)对不同pt-c6c10进行表征。
在核磁光谱仪上以400mhz记录1h核磁共振(nmr)光谱,并将四甲基硅烷用作内标。如图1所示,图1为对格氏反应后的中间体进行酸猝灭后进行的核磁测试得到的核磁谱图,从谱图中芳香区位置积分峰大小可以看出,中间体包含两种不同的单体,分别是噻吩基团上的一位取代b(1-mgbr),占总量的20%,和噻吩基团上的五位取代a(5-mgbr),占总量的80%。由于1-mgbr旁边有庞大的烷基侧链存在,位阻效应较大,因此只有5-mgbr可以进行下一步的催化转移缩聚合反应,这解释了该反应产率较低的原因,但同时保证了聚合物共轭骨架的区域规整性。如图2所示,为通过改变单体与催化剂的投料比,获得的不同分子量的pt-c6c10的1h核磁谱图,通过对谱图中芳香区位置积分峰进行积分,可推断出聚合物的大概分子量,同时从谱图中可以看出聚合物是比较纯的,说明使用制备型高效液相色谱(phplc)进行提纯能够获得较高纯度的pt-c6c10。
(2)采用配有两个tsk凝胶色谱柱(supermultiporehz-m)和uv检测器(254nm)的toshogpc系统(hlc-8320gpcecosec)在thf中进行凝胶渗透色谱,对不同pt-c6c10的分子量进行表征。
聚合物样品的分子量和多分散指数(pdi)是基于聚苯乙烯校准计算的。测得的gpc保留时间曲线如图3所示,测得聚合物的数均分子量(mn)、重均分子量(mw)及多分散指数(pdi)信息表如表2所示。从表中可以看出,随着单体与催化剂的投料比的增加,聚合物的分子量也随之增加,达到了控制聚合物的分子量的目的。在不同的单体与催化剂比例下,由聚合物体系获得的聚合度不符合活性聚合的基本规律,这可能是由于聚合过程中存在大量的热聚合反应所致。在加热系统中,随着单体量的增加,分子量也增加,表明热聚合反应增加。同时,由上述格氏反应结束后,只有80%(5-mgbr)的中间体可以进行下一步的聚合反应,这意味着在反应过程中部分单体会丢失,导致聚合物分子量下降。
表2不同单体/催化剂摩尔比的pt-c6c10分子量(mn,gpc)信息表
[a]为由gpc测得的聚合物的数均分子量;
[b]为由gpc测得的聚合物的重均分子量;
[c]为聚合物分散性指数,用于描述聚合物分子量分布;
[d]为聚合物的聚合度。
(3)采用差示扫描量热仪(日立高新技术科学dsc-7000x)测试pt-c6c10的玻璃化转变温度。
在液氮冷却、氮气流条件下以10℃min-1的冷却或加热速率,测得聚合物的玻璃化转变温度如图4(a,b)所示,为不同分子量的聚合物在第一次降温阶段(图4(a))和第二次升温阶段(图4(b))过程中,分别以不同的积分位置(起点,拐点和偏移)来确定的玻璃化转变温度(tg),具体的温度如表3所示,其中图4显示了pt-c6c10具有宽的玻璃态转变,这可能是由于围绕共轭骨架的聚合物周围存在大的烷基侧链。在降温或升温过程中,物体需要花费较长的时间(松弛时间)才能通过平衡状态下的聚合物运动(松弛或纠缠)转变为与外场兼容的新平衡状态(纠缠或松弛)。表3显示了在降温和升温阶段由起点,拐点和偏移确定的tg。经过两次dsc测试后,误差范围保持在1℃以内,表明该测试是可重复的,其中,tg,flex[a]表示由拐点确定的tg,tg,onset[b]表示由开始时间确定的tg,tg,offset[c]表示由结束时间确定的tg。
表3不同mn,的pt-c6c10的玻璃化转变温度表
(4)采用flory-fox、fox-loshaek、ogawa三种不同的数学模型来模拟pt-c6c10的tg随分子量增加的变化情况,以解释tg分布的合理性及验证该聚合物tg变化规律。
如表4所示,表4为pt-c6c10的数均分子量(mn,gpc)与玻璃化转变温度(tg)之间关系的flory-fox、fox-loshaek、ogawa方程模型信息表。通过使用三个不同的表达式,三个过程中的升温阶段具有更好的拟合关系。在六个不同的降温回归方程中r2为0.69、0.76、0.76、0.87、0.64、0.69,升温回归方程中的并且r2为0.96、0.96、0.96、0.94、0.94、0.96。升温回归方程中flory-fox模型的相关系数r2为0.96,这可能是因为聚合物在升温阶段开始时处于混合相状态(凝固相和熔融相),并且与降温阶段相比,具有较短的弛豫时间以达到另一个稳定状态。因此,通过升温确定tg可以获得更好的回归曲线。其中,flory-fox模型数学表达式:
表4采用方程模型拟合的pt-c6c10玻璃化转变温度信息表
[a]为当聚合物分子量趋于无限大时的玻璃化转变温度;
[b]每一种聚合物的特征常数;
[c]相关系数
图5(a,b)所示的是pt-c6c10的分子量(mn,gpc)与tg之间的方程模型关系图;其中,(a)为flory-fox和fox-loshaek方程模型关系图,(b)为ogawa方程模型关系图。图5(a)表明tg随着mn,gpc的增加而增加,但是当mn,gpc增加到一定程度时,tg趋于一定值。首先,随着mn,gpc的增加,tg逐渐增加,因为分子量的增加导致分数链增长,同时导致侧链增加。侧链的产生将阻碍链的旋转,从而增加tg值。然后,tg趋于一定值。这可能是由于分子链两端的末端链具有更大的迁移率。随着分子量的增加,端链的贡献逐渐降低,因此其对tg的影响也降低。
综上所述,本发明制备的粘弹性荧光共轭聚合物中,以噻吩基团作为共轭骨架,噻吩基团的侧链上具有两条较长的碳支链,形成庞大的侧链基团。这些侧链基团作为“增塑剂”,使共轭聚合物在室温下具有粘弹性,且在室温无溶剂状态下展现出了优异的延展性。其制备方法反应温度低,反应时间短,且通过改变单体和催化剂的用量比例可调整共轭聚合物聚合物分子量的大小,以实现对共轭聚合物的柔性程度的调节,从而适应不同领域对柔性共轭聚合物的需求。同时,由于噻吩基团上的烷基侧链基团具有较大的位阻效应,在催化转移缩聚反应过程中保证了合物共轭骨架的区域规整性,提高了共轭聚合物的荧光特性。本发明提供的粘弹性荧光共轭聚合物的制备方法简单易实现,能耗较小,制备的粘弹性荧光共轭聚合物可应用于柔性发光器件或柔性可穿戴设备中。
应当理解的是,本发明的应用不限于上述的举例,对本领域普通技术人员来说,可以根据上述说明加以改进或变换,所有这些改进和变换都应属于本发明所附权利要求的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!