合成方法和中间体与流程
合成方法和中间体
1.相关申请的交叉引用
2.本专利申请要求2019年12月20日提交的美国申请序列号62/951,836的优先权,所述申请以引用方式并入本文中。
背景技术:
3.乙型肝炎病毒(hbv)为嗜肝dna病毒家族的成员。人类感染hbv可引起肝脏的感染性炎性疾病。受感染的个体可能多年不会表现出症状。据估计,约有三分的一的世界人口在其生命中的某个时刻受到感染,其中包括3.5亿慢性携带者。
4.丁型肝炎病毒(hdv)是一种小的环状包膜rna病毒,其仅可在乙型肝炎病毒(hbv)的存在下繁殖。特别地,hdv需要hbv表面抗原蛋白进行自身繁殖。与hbv单独感染相比,hbv与hdv同时感染会导致更严重的并发症。与乙型肝炎病毒组合,丁型肝炎是所有肝炎感染中死亡率最高的。
5.国际专利申请公开号wo 2018/191278描述了可用于将sirna靶向肝的适合用于治疗例如hbv和/或hdv的缀合物。当前,需要可以用于制备此类缀合物的合成方法和合成中间体。
技术实现要素:
6.在一个方面,本发明提供了可以用于制备治疗性缀合物的合成方法和合成中间体化合物。
7.本发明还提供了通过施用由本发明的方法制备的治疗性缀合物来治疗人类的hbv和/或hdv感染的方法。
8.本发明还提供了用于治疗人类受试者的hbv和/或hdv感染的方法,该方法包括向该人类受试者施用治疗有效量的通过本发明的方法制备的治疗性缀合物,以及可用于治疗hbv和/或hdv的第二治疗剂。
9.本发明还提供了通过本发明的方法制备的化合物。
10.本发明还提供了通过本发明的方法制备的治疗性缀合物,其用于医学治疗。
11.本发明还提供了通过本发明的方法制备的治疗性缀合物,其任选地与另一种治疗剂组合用于hbv和/或hdv的预防性或治疗性治疗。
12.本发明还提供了通过本发明的方法制备的治疗性缀合物在制备任选地与另一种治疗剂组合用于治疗hbv和/或hdv的药物中的用途。
具体实施方式
13.除非另有说明,否则使用以下定义。
14.除非另有说明,否则术语“烷基”就其本身或作为另一个取代基的一部分时意指具有所示碳原子数目的直链或支链烃基(即,c
1-8
意指一至八个碳)。实例包括(c
1-c8)烷基、(c
2-c8)烷基、(c
1-c6)烷基、(c
2-c6)烷基和(c
3-c6)烷基。烷基的实例包括甲基、乙基、正丙基、
异丙基、正丁基、叔丁基、异丁基、仲丁基、正戊基、正己基、正庚基、正辛基和更高的同系物和异构体。
15.如本文所用,术语“保护基”是指通常用于阻隔或保护化合物上的特定官能团的取代基。例如,“氨基保护基”为连接于氨基的阻隔或保护化合物中的氨基官能性的取代基。合适的氨基保护基包括乙酰基、三氟乙酰基、叔丁氧羰基(boc)、苄氧羰基(cbz)和9-芴甲氧羰基(fmoc)。类似地,“羟基保护基”是指阻隔或保护羟基官能性的羟基的取代基。合适的保护基包括乙酰基和硅基。“羧基保护基”是指阻隔或保护羧基官能性的羧基的取代基。常见羧基保护基包括苯基磺酰基乙基、氰乙基、2-(三甲基硅基)乙基、2-(三甲基硅基)乙氧基甲基、2-(对甲苯磺酰基)乙基、2-(对硝基苯基亚磺酰基)乙基、2-(二苯基膦基)-乙基、硝基乙基等等。对于保护基和其使用的一般描述,参见p.g.m.wuts和t.w.greene,greene's protective groups in organic synthesis第4版,wiley-interscience,new york,2006。
16.如本文所用,在化学结构中与键交叉的波形线表示化学结构中波形键与分子的其余部分交叉的键的连接点。
17.当本文中的化合物式中的键以非立体化学方式(例如,扁平的)画出时,该键所连接的原子包括所有立体化学可能性。当本文中的化合物式中的键以经定义的立体化学方式(例如粗体、粗体-楔形、虚线或虚线-楔形)画出时,应理解除非另外指示,否则立体化学键所连接的原子富集于所描绘的绝对立体异构体。在一个实施方案中,该化合物可为至少51%所描绘的绝对立体异构体。在另一个实施方案中,该化合物可为至少60%所描绘的绝对立体异构体。在另一个实施方案中,该化合物可为至少80%所描绘的绝对立体异构体。在另一个实施方案中,该化合物可为至少90%所描绘的绝对立体异构体。在另一个实施方案中,该化合物可为至少95所描绘的绝对立体异构体。在另一个实施方案中,该化合物可为至少99%所描绘的绝对立体异构体。
18.衣壳抑制剂
19.如本文所述,术语“衣壳抑制剂”包括能够直接或间接地抑制衣壳蛋白的表达和/或功能的化合物。例如,衣壳抑制剂可包括但不限于抑制衣壳组装、诱导非衣壳聚合物的形成、促进过度的衣壳组装或误导的衣壳组装、影响衣壳稳定性和/或抑制rna的衣壳化的任何化合物。衣壳抑制剂还包括抑制复制过程内的一个或多个下游事件(例如,病毒dna合成、松弛环状dna(rcdna)向核中的转运、共价闭合环状dna(cccdna)形成、病毒成熟、出芽和/或释放等等)中的衣壳功能的任何化合物。例如,在某些实施方案中,该抑制剂可侦测地抑制衣壳蛋白的表达水平或生物活性,如例如使用本文所述的测定所测量的。在某些实施方案中,该抑制剂将rcdna和病毒生命周期的下游产物的水平抑制了至少5%、至少10%、至少20%、至少50%、至少75%或至少90%。
20.术语衣壳抑制剂包括国际专利申请公开号wo2013006394、wo2014106019和wo2014089296中描述的化合物,包括以下化合物:
21.22.术语衣壳抑制剂还包括化合物bay-41-4109(参见国际专利申请公开号wo/2013/144129)、at-61(参见国际专利申请公开号wo/1998/33501;和king,rw等人,antimicrob agents chemother.,1998,42,12,3179
–
3186)、dvr-01和dvr-23(参见国际专利申请公开号wo 2013/006394;和campagna,mr等人,j.of virology,2013,87,12,6931),和其药学上可接受的盐:
[0023][0024]
术语衣壳抑制剂还包括:
[0025][0026]
和其药学上可接受的盐。
[0027]
sag分泌抑制剂/rna去稳定剂
[0028]
如本文所述,术语“sag分泌抑制剂”包括能够直接或间接抑制携带sag(s、m和/或l表面抗原)的亚病毒颗粒和/或含dna的病毒颗粒自hbv感染细胞分泌的化合物。如本文所用,“sag分泌抑制剂”还称为“rna去稳定剂”,并且这些术语可互换使用。例如,在某些实施方案中,抑制剂可侦测地抑制sag的分泌,如例如使用本领域已知的或本文所述的测定(例如,elisa测定或通过蛋白质印迹法)所测量的。在某些实施方案中,抑制剂将sag的分泌抑制至少5%、至少10%、至少20%、至少50%、至少75%、或至少90%。在某些实施方案中,抑制剂将患者血清sag水平降低至少5%、至少10%、至少20%、至少50%、至少75%、或至少90%。
[0029]
术语sag分泌抑制剂包括在美国专利号8,921,381中描述的化合物,以及在美国专利申请公开号2015/0087659和2013/0303552中描述的化合物。例如,该术语包括化合物pbhbv-001和pbhbv-2-15和其药学上可接受的盐:
[0030][0031]
本发明的具体实施方案描述于下文中。
[0032]
在一个实施方案中,本发明提供了一种用于制备式1的化合物的方法:
[0033][0034]
其包括在40℃或更高的温度下,使式1-1的化合物:
[0035][0036]
与式1-2的化合物:
[0037][0038]
反应以提供式1的化合物。该反应可在纯净条件下或在一种或多种溶剂的存在下进行。在一个实施方案中,本发明在极性非质子溶剂诸如例如四氢呋喃、1,2-二氯乙烯、甲基四氢呋喃、甲苯、乙腈、二甲氧基乙烷或四氯化碳中进行。在一个实施方案中,反应在约0℃至约100℃范围内的温度下进行。在另一个实施方案中,反应在60℃或更高的温度下进行。在另一个实施方案中,反应在约60℃至约80℃范围内的温度下进行。
[0039]
在一个实施方案中,本发明提供了一种用于制备化合物3的结晶形式的方法:
[0040][0041]
其包括在转化期间不使用柱色谱法的情况下,将式1的化合物:
[0042][0043]
转化为化合物3的结晶形式。在一个实施方案中,该化合物可从包含二氯甲烷或乙
酸乙酯的溶剂中结晶。在另一个实施方案中,化合物从二氯甲烷或乙酸乙酯中结晶。
[0044]
在一个实施方案中,本发明提供了化合物3的结晶形式:
[0045][0046]
在一个实施方案中,本发明提供了一种用于制备其中r9为任选取代的苄氧羰基的式9的化合物的方法:
[0047][0048]
该方法包括将式8的化合物:
[0049][0050]
或其盐转化为式9的化合物。转化可在任何合适的温度下进行,并且可在纯净条件下或在一种或多种溶剂的存在下进行。在本发明的一个实施方案中,转化在极性或非极性非质子溶剂诸如例如二氯甲烷、氯仿、四氢呋喃、甲基四氢呋喃、四氯化碳、乙腈、吡啶、二甲基甲酰胺、二甲基乙酰胺或甲苯中进行。在一个实施方案中,转化在约0℃至约100℃范围内的温度下进行。在另一个实施方案中,转化在约15℃至约25℃范围内的温度下进行。在一个实施方案中,r9为苄氧羰基或硝基苄氧羰基。在一个实施方案中,通过在合适的碱的存在下在合适的溶剂中用苄氧羰基氯处理式8的化合物,将式8的化合物转化为式9的化合物。在一个实施方案中,碱为胺碱,诸如例如三甲胺、三乙胺、吡啶、二甲基氨基吡啶、二异丙基乙胺或三丙胺。
[0051]
在一个实施方案中,本发明提供了一种用于制备其中r9为任选取代的苄氧羰基的式10的化合物的方法:
[0052][0053]
该方法包括将对应式9的化合物:
[0054][0055]
转化为式10的化合物。转化可在任何合适的温度下进行,并且可在纯净条件下或在一种或多种溶剂的存在下进行。在一个实施方案中,该转化提供了至少约85%、90%或95%的β-异构体形式的式10的化合物。在本发明的一个实施方案中,转化在非极性非质子溶剂诸如例如二氯乙烷、二氯甲烷、乙腈、甲基四氢呋喃、四氢呋喃、二甲氧基乙烷或甲苯中进行。在一个实施方案中,转化在约0℃至约100℃范围内的温度下进行。在另一个实施方案
中,转化在约80℃至约85℃范围内的温度下进行。在另一个实施方案中,转化在约35℃至约45℃范围内的温度下进行。在另一个实施方案中,转化在约45℃至约55℃范围内的温度下进行。在另一个实施方案中,转化在约55℃至约65℃范围内的温度下进行。在另一个实施方案中,转化在最优化产物的β:α比例的温度下进行。在一个实施方案中,r9为苄氧羰基或硝基苄氧羰基。在一个实施方案中,通过在合适的催化剂和合适的溶剂的存在下用式7的化合物:
[0056][0057]
处理,将式9的化合物转化为式10的化合物。在一个实施方案中,催化剂为sc(otf)3、三氟甲磺酸三甲基硅基酯、氯化锌或4a分子筛。
[0058]
在一个实施方案中,本发明提供了一种用于制备其中r9为任选取代的苄氧羰基的式10的化合物的方法:
[0059][0060]
该方法包括将式8的化合物:
[0061][0062]
或其盐转化为对应式9的化合物;
[0063][0064]
并且随后在不通过色谱法纯化式9的化合物的情况下,将对应式9的化合物转化为式10的化合物。
[0065]
在一个实施方案中,本发明提供了一种用于制备式11的盐的方法:
[0066][0067]
其包括在合适的催化剂的存在下和在合适的溶剂的存在下用氢气和三氟乙酸处理其中r9为任选取代的苄氧羰基的式10的化合物:
[0068][0069]
在一个实施方案中,合适的催化剂包括钯碳。在一个实施方案中,合适的溶剂包括四氢呋喃。该反应可在任何合适的温度下进行。在一个实施方案中,反应在约0℃至约50℃
范围内的温度下进行。在另一个实施方案中,反应在约20℃至约25℃范围内的温度下进行。在一个实施方案中,r9为苄氧羰基或硝基苄氧羰基。
[0070]
在一个实施方案中,本发明提供了一种用于制备式15d的化合物或其盐的方法:
[0071][0072]
其包括将其中每个r
15
为(c
1-c6)烷基的式15c的化合物:
[0073][0074]
转化为式15d的化合物或其盐。转化可在任何合适的温度下进行,并且可在纯净条件下或在一种或多种溶剂的存在下进行。在本发明的一个实施方案中,转化在极性质子溶剂诸如例如甲醇、乙醇、四氢呋喃和/或水中进行。在一个实施方案中,转化在约0℃至约100℃范围内的温度下进行。在另一个实施方案中,转化在约15℃至约25℃范围内的温度下进行。在一个实施方案中,转化在合适的碱诸如例如氢氧化钠、氢氧化锂或氢氧化钾的存在下进行。
[0075]
在一个实施方案中,本发明提供了一种用于制备其中每个r
15
为(c
1-c6)烷基的式15c的化合物的方法:
[0076][0077]
其包括使式15a的化合物:
[0078][0079]
或其盐与对应式15b的化合物:
[0080][0081]
或其盐反应,以提供式15c的化合物。该反应可在任何合适的温度下进行,并且可在纯净条件下或在一种或多种溶剂的存在下进行。在本发明的一个实施方案中,反应在极性非质子溶剂诸如例如二甲基甲酰胺、二氯甲烷、1,2-二氯乙烷或二甲基乙酰胺中进行。在一个实施方案中,反应在约0℃至约50℃范围内的温度下进行。在另一个实施方案中,反应在约5℃至约10℃范围内的温度下进行。在一个实施方案中,反应在合适的碱的存在下进行。在一个实施方案中,碱为受阻胺碱,诸如例如二异丙基乙胺、三甲胺、吡啶或二甲基氨基吡啶。在一个实施方案中,反应在合适的偶联剂诸如例如1-乙基-3-(3-二甲基氨基丙基)碳二亚胺edc、n,n'-二环己基-碳二亚胺dcc、(1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑并
[4,5-b]吡啶鎓3-氧化物六氟磷酸盐hatu、(2-(1h-苯并三唑-1-基)-1,1,3,3-四甲基脲六氟磷酸盐hbtu或丙烷膦酸酐t3p)的存在下进行。
[0082]
在一个实施方案中,本发明提供了一种用于制备其中每个r
15
为(c
1-c6)烷基的式13a的化合物的方法:
[0083][0084]
其包括将其中每个r
15
为(c
1-c6)烷基的对应式15c的化合物:
[0085][0086]
转化为式13a的化合物。转化可在任何合适的温度下进行,并且可在纯净条件下或在一种或多种溶剂的存在下进行。在本发明的一个实施方案中,转化在极性质子溶剂诸如例如甲醇、乙酸乙酯、四氢呋喃、甲基四氢呋喃或乙醇中进行。在一个实施方案中,转化在约0℃至约100℃范围内的温度下进行。在另一个实施方案中,转化在约15℃至约25℃范围内的温度下进行。在一个实施方案中,转化在合适的催化剂诸如例如钯碳或pd(oh)2的存在下进行。
[0087]
在一个实施方案中,本发明提供了一种用于制备其中每个r
15
为(c
1-c6)烷基并且t为任选取代的三苯基甲基的式13b的化合物的方法:
[0088][0089]
其包括将对应式13a的化合物:
[0090][0091]
转化为式13b的化合物。转化可在任何合适的温度下进行,并且可在纯净条件下或在一种或多种溶剂的存在下进行。在本发明的一个实施方案中,转化在非极性非质子溶剂诸如例如二氯甲烷、1,2-二氯乙烷、二甲基甲酰胺或二甲基乙酰胺中进行。在一个实施方案中,转化在约-78℃至约100℃范围内的温度下进行。在另一个实施方案中,转化在约0℃至约30℃范围内的温度下进行。在一个实施方案中,该转化在合适的偶联剂诸如例如1-乙基-3-(3-二甲基氨基-丙基)碳二亚胺edc、n,n'-二环己基碳二亚胺dcc、(1-[双(二甲氨基)-亚甲基]-1h-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸盐hatu、(2-(1h-苯并三唑-1-基)-1,1,3,3-四甲基脲六氟磷酸盐hbtu或丙烷膦酸酐t3p)的存在下进行。在一个实施方案中,通过在合适的酰胺形成条件下,用其中dmtr为4,4-二甲氧基三苯基甲基的对应式6的化
合物:
[0092][0093]
或其盐处理式13a的化合物,将式13a的化合物转化为式13b的化合物。在一个实施方案中,在1-乙基-3-(3-二甲基氨基丙基)碳二亚胺的存在下,在约0℃至约30℃范围内的温度下,在二氯甲烷中,将式13a的化合物用下式的化合物:
[0094][0095]
处理。
[0096]
在一个实施方案中,本发明提供了一种用于制备式13c的化合物的方法:
[0097][0098]
其包括将其中每个r
15
为(c
1-c6)烷基并且t为任选取代的三苯基甲基的式13b的化合物:
[0099][0100]
转化为式13c的化合物。转化可在任何合适的温度下进行,并且可在纯净条件下或在一种或多种溶剂的存在下进行。在本发明的一个实施方案中,转化在极性质子溶剂诸如例如甲醇、乙醇、四氢呋喃和/或水中进行。在一个实施方案中,转化在约0℃至约100℃范围内的温度下进行。在另一个实施方案中,转化在约20℃至约40℃范围内的温度下进行。在一个实施方案中,转化在合适的碱诸如例如氢氧化钾、氢氧化锂或氢氧化钠的存在下进行。在一个实施方案中,通过在包含甲醇和水的溶剂中用氢氧化钾处理,将式13b的化合物转化为式13c的化合物。
[0101]
在一个实施方案中,本发明提供了一种用于制备式13cc的化合物的结晶钾盐的方法:
[0102][0103]
其包括在甲醇中用氢氧化钾处理式13cc的化合物或其盐。在一个实施方案中,式13cc的化合物的结晶钾盐可如实施例30中所述制备。
[0104]
在一个实施方案中,本发明提供了一种用于制备式11b的化合物的方法:
[0105][0106]
其包括将式11a的化合物:
[0107][0108]
或其盐转化为式11b的化合物。转化可在任何合适的温度下进行,并且可在纯净条件下或在一种或多种溶剂的存在下进行。在本发明的一个实施方案中,转化在非极性非质子溶剂诸如例如二氯甲烷、1,2-二氯乙烷、甲基四氢呋喃、四氢呋喃、二甲基甲酰胺或二甲基乙酰胺中进行。在一个实施方案中,转化在约0℃至约100℃范围内的温度下进行。在另一个实施方案中,转化在约5℃至约30℃范围内的温度下进行。在一个实施方案中,通过在二氯甲烷中用1-乙基-3-(3-二甲基氨基丙基)碳二亚胺edc、n,n'-二环己基碳二亚胺dcc、(1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸盐hatu、(2-(1h-苯并三唑-1-基)-1,1,3,3-四甲基脲六氟磷酸盐hbtu或丙烷膦酸酐t3p)处理式11a的化合物或其盐,将式11a的化合物转化为式11b的化合物。
[0109]
在一个实施方案中,本发明提供了一种用于制备式12的化合物的方法:
[0110][0111]
其包括将式11b的化合物:
[0112][0113]
转化为式12的化合物。转化可在任何合适的温度下进行,并且可在纯净条件下或在一种或多种溶剂的存在下进行。在本发明的一个实施方案中,转化在非极性非质子溶剂诸如例如,二氯甲烷、1,2-二氯乙烷、甲基四氢呋喃、四氢呋喃、二甲基甲酰胺或二甲基乙酰胺中进行。在一个实施方案中,转化在约0℃至约50℃范围内的温度下进行。在另一个实施方案中,转化在约0℃至约30℃范围内的温度下进行。在一个实施方案中,转化在合适的碱的存在下进行。在一个实施方案中,该碱为受阻胺碱,诸如例如二异丙基乙胺、三甲胺、二甲基氨基吡啶或吡啶。在一个实施方案中,通过在合适的碱和合适的溶剂的存在下,用式11的化合物:
[0114][0115]
或其盐处理式11b的化合物,将式11b的化合物转化为式12的化合物。在一个实施方案中,通过在二异丙基乙胺和包含二氯甲烷的溶剂的存在下,用式11的化合物的三氟乙酸盐:
[0116][0117]
处理式11b的化合物,将式11b的化合物转化为式12的化合物。
[0118]
在一个实施方案中,本发明提供了一种用于制备式13的化合物或其盐的方法:
[0119][0120]
其包括将式12的化合物:
[0121][0122]
还原以提供式13的化合物或其盐。还原可在任何合适的温度下进行,并且可在纯净条件下或在一种或多种溶剂的存在下进行。在本发明的一个实施方案中,还原在极性非质子溶剂诸如例如四氢呋喃、甲基四氢呋喃或乙酸乙酯中进行。在一个实施方案中,还原在约0℃至约50℃范围内的温度下进行。在另一个实施方案中,还原在约0℃至约30℃范围内的温度下进行。在一个实施方案中,还原在合适的催化剂诸如例如钯碳的存在下进行。在一个实施方案中,式13的化合物或其盐为下式的三氟乙酸盐:
[0123][0124]
在一个实施方案中,本发明提供了一种用于制备式14的化合物的方法:
[0125][0126]
其包括将式13的化合物:
[0127][0128]
或其盐转化为式14的化合物。转化可在任何合适的温度下进行,并且可在纯净条件下或在一种或多种溶剂的存在下进行。在本发明的一个实施方案中,转化在非极性非质子溶剂诸如例如二氯甲烷、1,2-二氯乙烷、甲基四氢呋喃、四氢呋喃、二甲基甲酰胺或二甲基乙酰胺中进行。在一个实施方案中,转化在约-78℃至约25℃范围内的温度下进行。在另
一个实施方案中,转化在约-25℃至约30℃范围内的温度下进行。在一个实施方案中,转化在合适的碱的存在下进行。在一个实施方案中,碱为胺碱,诸如例如三甲胺、三乙胺、二异丙基乙胺、二甲基氨基吡啶、吡啶或三丙胺。在一个实施方案中,转化在合适的偶联剂诸如例如丙烷膦酸酐的存在下进行。在一个实施方案中,通过在约-15℃至约15℃范围内的温度下,在偶联剂诸如例如1-乙基-3-(3-二甲基氨基丙基)碳二亚胺edc、(1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸盐hatu、(2-(1h-苯并三唑-1-基)-1,1,3,3-四甲基脲六氟磷酸盐hbtu或丙烷膦酸酐t3p的存在下,在包含二氯甲烷的溶剂中,用下式的化合物:
[0129][0130]
或其盐来处理式13的化合物,将式13的化合物转化为式14的化合物。
[0131]
在一个实施方案中,本发明提供了一种用于制备其中r
16
为胺保护基的式16的化合物的方法:
[0132][0133]
其包括将式13的化合物:
[0134][0135]
或其盐转化为式16的化合物。转化可在任何合适的温度下进行,并且可在纯净条件下或在一种或多种溶剂的存在下进行。在本发明的一个实施方案中,转化在非极性非质子溶剂诸如例如二氯甲烷、1,2-二氯乙烷、甲基四氢呋喃、四氢呋喃、二甲基甲酰胺或二甲基乙酰胺中进行。在一个实施方案中,转化在约-78℃至约50℃范围内的温度下进行。在另一个实施方案中,转化在约-25℃至约50℃范围内的温度下进行。在一个实施方案中,转化在合适的碱的存在下进行。在一个实施方案中,碱为胺碱,诸如例如三甲胺、三乙胺或三丙
胺、二异丙基乙胺、二甲基氨基吡啶或吡啶。在一个实施方案中,转化在合适的偶联剂诸如例如1-乙基-3-(3-二甲基氨基丙基)碳二亚胺edc、(1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸盐hatu、(2-(1h-苯并三唑-1-基)-1,1,3,3-四甲基脲六氟磷酸盐hbtu、或丙烷膦酸酐t3p的存在下进行。在一个实施方案中,通过在合适的偶联条件下,用其中r
16
为胺保护基的对应式15dd的化合物:
[0136][0137]
或其盐处理式13的化合物,将式13的化合物或其盐转化为式16的化合物。在一个实施方案中,在合适的偶联条件下,将式13的化合物的三氟乙酸盐:
[0138][0139]
用其中r
16
为苄氧羰基的式15d的化合物处理:
[0140][0141]
以提供其中r
16
为苄氧羰基的式16的化合物。在一个实施方案中,在丙烷膦酸酐、三甲胺和包含二氯甲烷的溶剂的存在下,用式15d或15dd的化合物处理式13的化合物,以提供式16的化合物。
[0142]
在一个实施方案中,本发明提供了一种用于制备其中r
18
为合适的保护基的
[0143]
式18的化合物的方法:
[0144][0145]
其包括将式13的化合物:
[0146][0147]
或其盐转化为式18的化合物。转化可在任何合适的温度下进行,并且可在纯净条件下或在一种或多种溶剂的存在下进行。在本发明的一个实施方案中,转化在非极性非质子溶剂诸如例如二氯甲烷、1,2-二氯乙烷、甲基四氢呋喃、四氢呋喃、二甲基甲酰胺或二甲基乙酰胺中进行。在一个实施方案中,转化在约-78℃至约50℃范围内的温度下进行。在另一个实施方案中,转化在约-25℃至约50℃范围内的温度下进行。在一个实施方案中,转化在合适的碱的存在下进行。在一个实施方案中,碱为胺碱,诸如例如三甲胺、三乙胺或三丙胺、二异丙基乙胺、二甲基氨基吡啶或吡啶。在一个实施方案中,转化在合适的偶联剂诸如例如1-乙基-3-(3-二甲基氨基丙基)碳二亚胺edc、(1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸盐hatu、(2-(1h-苯并三唑-1-基)-1,1,3,3-四甲基脲六氟磷酸盐hbtu、或丙烷膦酸酐t3p的存在下进行。在一个实施方案中,通过在合适的偶联条件下,用其中r
18
为合适的保护基的式13ccc的化合物:
[0148][0149]
或其盐处理式13的化合物或其盐,将式13的化合物或其盐转化为式18的化合物。在一个实施方案中,在合适的偶联条件下,式13的化合物的三氟乙酸盐:
[0150][0151]
用其中r
18
为4,4-二甲氧基三苯基甲基的式13ccc的化合物处理,以提供其中r
18
为4,4-二甲氧基三苯基甲基的式18的化合物:
[0152][0153]
在一个实施方案中,在丙烷膦酸酐、三甲胺和包含二氯甲烷的溶剂的存在下,用式13ccc的化合物处理式13的化合物,以提供式18的化合物。
[0154]
在一个实施方案中,本发明提供了一种用于制备式16-2的化合物的方法:
[0155][0156]
其包括将式16-1的化合物:
[0157][0158]
或其盐转化为式16-2的化合物。转化可在任何合适的温度下进行,并且可在纯净条件下或在一种或多种溶剂的存在下进行。在本发明的一个实施方案中,转化在非极性非质子溶剂诸如例如二氯甲烷、1,2-二氯乙烷、氯仿或四氯化碳中进行。在一个实施方案中,转化在约-78℃至约100℃范围内的温度下进行。在另一个实施方案中,转化在约-0℃至约30℃范围内的温度下进行。在一个实施方案中,通过活化式16-1的化合物中的羧酸基团,例如通过用草酰氯处理式16-1的化合物,并用叔丁醇处理所得的羧酸氯基团来进行转化,以提供式16-2的化合物。
[0159]
在一个实施方案中,本发明提供了一种用于制备式16-3的化合物的方法:
[0160][0161]
其包括将式16-2的化合物:
[0162][0163]
转化为式16-3的化合物。转化可在任何合适的温度下进行,并且可在纯净条件下或在一种或多种溶剂的存在下进行。在本发明的一个实施方案中,转化在极性质子溶剂诸如例如甲醇或乙醇中进行。在一个实施方案中,反应在约-78℃至约50℃范围内的温度下进行。在另一个实施方案中,转化在约-0℃至约50℃范围内的温度下进行。在一个实施方案中,转化在合适的催化剂诸如例如钯碳的存在下进行。
[0164]
在一个实施方案中,本发明提供了一种用于制备式16-4的化合物的方法:
[0165][0166]
其包括将式16-3的化合物:
[0167][0168]
转化为式16-4的化合物。转化可在任何合适的温度下进行,并且可在纯净条件下或在一种或多种溶剂的存在下进行。在本发明的一个实施方案中,转化在非极性非质子溶剂诸如例如二氯甲烷、1,2-二氯乙烷、甲基四氢呋喃、四氢呋喃、二甲基甲酰胺或二甲基乙酰胺中进行。在一个实施方案中,转化在约-78℃至约50℃范围内的温度下进行。在另一个实施方案中,转化在约-0℃至约50℃范围内的温度下进行。在一个实施方案中,转化在合适的碱的存在下进行。在一个实施方案中,碱为胺碱,诸如例如三甲胺、三乙胺或三丙胺、二异丙基乙胺、二甲基氨基吡啶或吡啶。在一个实施方案中,转化在合适的偶联剂诸如例如1-乙基-3-(3-二甲基氨基丙基)碳二亚胺edc、(1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸盐hatu、(2-(1h-苯并三唑-1-基)-1,1,3,3-四甲基脲六氟磷酸盐hbtu、或丙烷膦酸酐t3p的存在下进行。
[0169]
在一个实施方案中,本发明提供了一种用于制备式16-5的化合物或其盐的方法:
[0170][0171]
其包括将式16-4的化合物:
[0172][0173]
转化为式16-5的化合物。转化可在任何合适的温度下进行,并且可在纯净条件下或在一种或多种溶剂的存在下进行。在本发明的一个实施方案中,转化在极性质子溶剂诸如例如甲醇、乙醇、四氢呋喃或乙酸乙酯中进行。在一个实施方案中,转化在约-78℃至约50℃范围内的温度下进行。在另一个实施方案中,转化在约-0℃至约50℃范围内的温度下进行。在一个实施方案中,转化在合适的催化剂诸如例如钯碳的存在下进行。
[0174]
在一个实施方案中,本发明提供了一种用于制备式16d的化合物或其盐的方法:
[0175][0176]
其包括将式16-5的化合物:
[0177][0178]
转化为式16d的化合物。转化可在任何合适的温度下进行,并且可在纯净条件下或
在一种或多种溶剂的存在下进行。在本发明的一个实施方案中,转化在非极性非质子溶剂诸如例如二氯甲烷、1,2-二氯乙烷、甲基四氢呋喃、四氢呋喃、二甲基甲酰胺或二甲基乙酰胺中进行。在一个实施方案中,转化在约-78℃至约50℃范围内的温度下进行。在另一个实施方案中,转化在约0℃至约50℃范围内的温度下进行。在一个实施方案中,转化在合适的碱的存在下进行。在一个实施方案中,碱为胺碱,诸如例如三甲胺、三乙胺或三丙胺、二异丙基乙胺、二甲基氨基吡啶或吡啶。在一个实施方案中,转化在合适的偶联剂诸如例如1-乙基-3-(3-二甲基氨基丙基)碳二亚胺edc、(1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸盐hatu、(2-(1h-苯并三唑-1-基)-1,1,3,3-四甲基脲六氟磷酸盐hbtu、或丙烷膦酸酐t3p的存在下进行。
[0179]
在一个实施方案中,本发明提供了一种用于制备式16e的化合物或其盐的方法:
[0180][0181]
其包括将式16d的化合物:
[0182][0183]
或其盐转化为式16d的化合物。转化可以在任何合适的温度下进行,并且可以在纯净条件下或在一种或多种溶剂的存在下进行。在本发明的一个实施方案中,转化在非极性非质子溶剂诸如例如二氯甲烷、氯仿或四氯化碳中进行。在一个实施方案中,转化在约-25℃至约50℃范围内的温度下进行。在另一个实施方案中,转化在约0℃至约50℃范围内的温
度下进行。在一个实施方案中,转化在合适的酸的存在下进行。在一个实施方案中,该酸为三氟乙酸。
[0184]
在一个实施方案中,本发明提供了一种用于制备式16的化合物或其盐的方法:
[0185][0186]
其包括将式16e的化合物:
[0187][0188]
或其盐转化为式16的化合物。转化可在任何合适的温度下进行,并且可在纯净条件下或在一种或多种溶剂的存在下进行。在本发明的一个实施方案中,转化在极性非质子溶剂诸如例如二甲基甲酰胺、二氯甲烷或二甲基氨基吡啶中进行。在一个实施方案中,转化在约-25℃至约25℃范围内的温度下进行。在另一个实施方案中,转化在约0℃至约10℃范围内的温度下进行。在一个实施方案中,转化在合适的碱的存在下进行。在一个实施方案中,该碱为受阻胺碱,诸如例如二异丙基乙胺、三甲胺、二甲基氨基吡啶或吡啶。在一个实施方案中,转化在合适的偶联剂诸如例如1-乙基-3-(3-二甲基氨基丙基)碳二亚胺edc的存在下进行。在一个实施方案中,转化在合适的羟基苯并三唑、n,n'-二环己基碳二亚胺dcc、(1-[双(二甲基氨基)-亚甲基]-1h-1,2,3-三唑并[4,5]-b]吡啶鎓3-氧化物六氟磷酸盐hatu或丙烷膦酸酐t3p)的存在下进行。在一个实施方案中,通过在合适的偶联条件下,使式16e的
化合物或其盐与式11的化合物:
[0189][0190]
或其盐反应,将式16e的化合物或其盐转化为式16的化合物或其盐。
[0191]
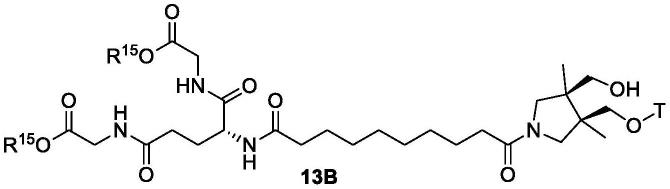
式16和式18的化合物可以用于制备治疗性缀合物,包括国际专利申请公开号wo 2018/191278中描述的对应治疗性缀合物。
[0192]
本发明现在将通过以下非限制性实施例来说明。
[0193]
实施例
[0194]
方案1
[0195][0196]
对于化合物1-6,粗体-楔形键表示顺式异构体,而不为绝对立体化学。本发明提供了具有两个顺式构型的化合物1-6。当将化合物1-6掺入到本文的其他化合物中时,来自化合物1-6中的粗体-楔形键表示顺式构型,而其中任何其他粗体、粗体-楔形、虚线或虚线-楔形键表示绝对立体化学。
[0197]
实施例1.化合物3的合成
[0198][0199]
向化合物1-1(200g,1.58mol)的2-methf(2.4l)溶液中添加三氟乙酸tfa(5.4g,4.7mmol)。将反应混合物加热至65-70℃至70℃,并且缓慢添加化合物1-2(414g,1.74mol),将反应温度维持在65-70℃。添加完成后,将反应混合物在65℃至70℃下加热不少于2小时,直到通过uplc确认反应完成(化合物1-1的消失)。然后将反应混合物冷却至-5至0℃。缓慢添加红铝(1.6kg,4.75mol,60-70%的甲苯溶液),将温度维持在低于-5至0℃。然后将反应混合物温热至25℃至30℃,并搅拌不少于12小时,直到通过uplc确认反应完成(化合物1-2的消失)。在另一个反应器2中,将10%naoh溶液(4.0l)冷却至0℃。通过将反应混合物转移
至冷10%naoh溶液中来淬灭反应混合物,同时将温度维持在低于30℃。转移完成后,将淬灭混合物搅拌3小时,然后使各层分离。分离有机层。用水(2.0l)、15%盐水(2.0l)洗涤有机层,并蒸发至干。呈浅黄色油的化合物2的粗残余物(424g)直接用于下一步骤。
[0200]
将化合物2(424g,1.7mol)溶于meoh(1.7l)中。添加活性炭(42.4g,0.10重量/重量),并加热到40-45℃持续2h。将热溶液经由hyflo床过滤,并用meoh(424ml)洗涤。将滤液转移至氢化高压釜烧瓶中,脱气并用n2吹扫两次。装入10重量%的pd/c(50%湿,42.4g),并将混合物脱气并用h2吹扫两次。将反应混合物在h2气氛(100psi)下搅拌不少于20小时,直到通过uplc确认反应完成。将混合物脱气并用n2吹扫,经由硅藻土垫过滤。将滤液蒸发至接近干燥,与乙酸乙酯(2x848ml)共蒸馏,在25至30℃下用乙酸乙酯(424ml)研磨3h。过滤固体,用冷乙酸乙酯(212ml)洗涤,并在《30℃下真空干燥。获得呈灰白色固体的化合物3(180.0g,71%)。m/z 160.11[m+h]
+
。1h nmr(400mhz,dmso-d6)δ4.18(s,2h),3.34(s,2h),3.25(d,j=10.5hz,2h),2.80(d,j=10.6hz,2h),2.44(d,j=10.5hz,2h),0.88(d,j=1.7hz,6h)。
[0201]
实施例2.化合物4的合成
[0202][0203]
在搅拌下将亚硫酰氯(100.9g,848mmol)和dmf(1.0g)依次装入到癸二酸甲酯(135.0g,848mmol)的dcm(1350ml)溶液中,同时将内部温度维持在20至30℃。将混合物在20℃至30℃下搅拌不少于2小时,直到通过uplc由癸二酸甲酯消失确认反应完成。将混合物蒸发至干,并将残余物与dcm(675ml)共蒸馏,然后将酰氯制成dcm(675ml)溶液。在另一个烧瓶中,将化合物3在dcm(675ml)、水(1350ml)、k2co3(239.0g,2544mmol)中的混合物冷却至0至5℃。向此冷混合物中,以15分钟的间隔分四批在搅拌下缓慢滴加dcm中的酰氯溶液,将温度维持在低于5℃。然后将反应混合物温热至25℃至30℃,并搅拌不少于12小时,直到通过uplc确认反应完成。将混合物用乙酸乙酯(1500ml)稀释,分离有机层,用水(1350ml)、盐水(675ml)洗涤,经na2so4(135g)干燥,蒸发并在《45℃下真空干燥。获得呈淡黄色液体的化合物4(260g,86%)。m/z 358.18[m+h]
+
。1h nmr(400mhz,dmso-d6)δ4.66(ddd,j=13.8,6.1,3.8hz,2h),3.51(dd,j=10.2,2.2hz,1h),3.41
–
3.32(m,1h),3.31(tt,j=7.6,3.8hz,4h),3.09(dd,j=10.3,2.3hz,1h),2.99(dd,j=11.9,2.4hz,1h),2.49(d,j=1.7hz,1h),2.27(td,j=7.4,2.4hz,2h),2.14(t,j=7.6hz,2h),1.55
–
1.40(m,4h),1.26
–
1.21(m,8h),1.01
–
0.90(m,6h)。
[0204]
实施例3.化合物5的合成
[0205][0206]
在20℃至30℃下,向化合物4(35.0g,9.8mmol)的dcm(350ml)溶液中添加三甲胺
tea(14.87g,14.7mmol)和dmap(1.2g,1.0mmol)。将此混合物冷却至0℃至5℃,并添加dmtrcl(33.2g,9.8mmol)。将反应混合物在相同温度下搅拌不少于2小时,直到通过uplc确认反应完成。添加水(350ml),并将混合物温热至20至30℃,并搅拌30分钟。分离水层,并用dcm(70ml)萃取。合并有机层,并用nahco3水溶液(350ml)、盐水(350ml)洗涤,经na2so4(35.0g)干燥,过滤并蒸发至干。粗残余物通过硅胶柱色谱法纯化(15至60%ea/己烷),得到呈浅黄色油的纯化合物5(35.0g,54.1%)。m/z 660.58[m+h]
+
。1h nmr(400mhz,氯仿-d)δ7.40(d,j=8.1hz,2h),7.34
–
7.17(m,9h),6.83(dd,j=8.8,2.6hz,4h),3.80(s,6h),3.67(s,3h),3.56(d,j=12.3hz,1h),3.49
–
3.18(m,3h),3.12(d,j=9.9hz,1h),3.04(s,1h),2.29(td,j=7.7,3.8hz,2h),2.17(q,j=6.2,4.8hz,2h),2.08
–
2.02(m,1h),1.30(s,12h),1.18(d,j=23.3hz,3h),1.03(d,j=4.8hz,3h)。
[0207]
实施例4.化合物6的合成
[0208][0209]
将化合物5(35.0g,5.3mmol)的meoh/水(1:1,700ml)溶液冷却至0℃。添加lioh.h2o(4.86g,11.6mmol),并将混合物搅拌不少于1小时,直到通过uplc确认反应完成。蒸发甲醇,添加水,并将混合物冷却至0至5℃。用磷酸二氢钠溶液将混合物中和至约ph 7.0,然后使用乙酸酸化至ph 6至6.5,同时将温度维持在低于5℃。用dcm(2x350ml)萃取含水混合物,并蒸发至完全干燥,然后在45℃下在真空烘箱中进一步干燥。获得呈灰白色固体的化合物6(28.3g,82%)。m/z 646.54[m+h]
+
。1h nmr(400mhz,dmso-d6)δ11.96(s,1h),7.32(p,j=7.6hz,4h),7.21(t,j=7.6hz,5h),6.87(d,j=8.2hz,4h),4.60
–
4.48(m,1h),3.72(s,6h),3.46(dd,j=30.2,11.0hz,1h),3.20(dd,j=25.3,11.0hz,1h),3.13
–
2.83(m,5h),2.12(dq,j=31.7,7.6hz,4h),1.49
–
1.41(m,4h),1.22(d,j=10.2hz,10h),1.11
–
0.99(m,4h)。
[0210]
方案2
[0211][0212]
实施例5.化合物9的合成
[0213][0214]
将与甲苯共沸浓缩以去除任何水内容物之后的19.0g(1.0当量)化合物8、和190ml(10v)的dcm装入到500ml反应器中。冷却至-20℃后,使用注射泵在-20至-10℃下缓慢装入
20.6g(0.95当量)的cbz-cl持续2小时。然后,使用注射泵在-20至-7℃下缓慢装入14.2g(1.10当量)的tea持续2小时。将反应混合物在室温搅拌17小时以完成反应转化。依次用95ml(5v)的1n hcl、95ml(5v)的8重量%nahco3和95ml(5v)盐水洗涤内容物。然后,将38ml(2v)的纯化水添加至有机层,并将内容物在65℃水浴下在20托下浓缩。与水进行共沸浓缩的过程重复两次(与水蒸馏去除杂质)。共沸浓缩后,将浓缩物用57ml(3v)的dcm稀释并用19g(1s)的na2so4处理。将内容物过滤并用38ml(2v)的dcm洗涤废弃物。将滤液在65℃水浴下在20托下浓缩,然后将其在50℃下在全真空下干燥一周末,得到呈无色油的产物化合物9(185g,97%产率)。m/z 284.2[m+h]
+
。1h nmr(600mhz,dmso-d6)δ7.39
–
7.28(m,3h),7.32
–
7.22(m,2h),5.73(s,1h),5.00(s,3h),4.56(t,j=5.5hz,2h),3.47(t,j=5.3hz,4h),3.40(td,j=5.6,5.2,2.2hz,7h),3.33(s,1h),3.14(q,j=6.0hz,4h)
[0215]
实施例6.化合物10的合成
[0216][0217]
向化合物9(10g,35.3mmol)的dce(100ml)溶液中添加化合物7(16.5g,42.4mmol)和tmsotf(0.6ml,3.5mmol)。将混合物在60至65℃下搅拌不少于3小时,直到通过uplc确认反应完成。使混合物冷却至20℃至25℃,并依次用8重量%的nahco3水溶液(2x60ml)、1n hcl(120ml)、盐水(120ml)洗涤,经na2so4(120g)干燥并蒸发至干,得到呈淡黄色糖浆的化合物10(22.7g,定量产率)。m/z 613.3[m+h]
+
。1h nmr(600mhz,dmso-d6)δ7.78(d,j=9.2hz,1h),7.38
–
7.26(m,5h),7.29
–
7.22(m,1h),5.20(d,j=3.4hz,1h),5.01
–
4.93(m,3h),4.54(d,j=8.5hz,1h),4.01(m,3h),3.86(m,1h),3.76(m,1h),3.60
–
3.51(m,1h),3.54
–
3.43(m,6h),3.39(t,j=6.0hz,2h),3.13(q,j=6.0hz,2h),2.08(s,3h),1.98(s,3h),1.87(s,3h),1.75(s,3h)。
[0218]
实施例7.化合物11的合成
[0219][0220]
向化合物10(110g,179mmol)的thf(100ml)溶液中添加tfa(20.5g,179mmol)。将混合物脱气并用n2吹扫两次。装入10重量%的pd/c(11g),并且将混合物脱气并用h2吹扫两次。使混合物在h2气氛(70psi)下搅拌不少于3小时,直到通过uplc确认反应完成。将混合物脱气并用n2吹扫,经由硅藻土垫过滤。蒸发滤液至完全干燥,得到呈淡黄色泡沫状固体的化合物11(106g,定量产率)。m/z 479.2[m+h]
+
。1h nmr(600mhz,dmso-d6)δ7.93(dd,j=12.4,5.3hz,4h),5.20(d,j=3.4hz,1h),4.96(dd,j=11.2,3.4hz,1h),4.54(d,j=8.5hz,1h),4.06
–
3.96(m,3h),3.88(dt,j=11.1,8.8hz,1h),3.78(m,1h),3.58(t,j=5.2hz,3h),3.58
–
3.45(m,6h),2.96(h,j=5.6hz,2h),2.08(s,3h),1.98(s,3h),1.87(s,3h),1.76(s,3h)。
[0221]
方案3
[0222][0223]
实施例8.化合物15c的合成
[0224][0225]
将dmf(1000ml)和dipea(275.3g,2.13mmol)的溶液冷却至0至5℃。依次装入化合物15-a(100g,0.35mol)、edc.hcl(217.1g,1.13mol)和hobt一水合物(173.9g,1.13mol),同时将温度维持在0至5℃。搅拌10分钟,然后装入化合物15-b(163.4g,1.17mol)。使反应混合物温热至25℃至30℃,并搅拌不少于16小时,直到通过uplc确认反应完成。通过缓慢添加乙醇(1000ml),之后添加水(4500ml)来稀释反应混合物,并在25℃至30℃下搅拌不少于4h。过滤出形成的沉淀,用水(1000ml)洗涤,并将固体在《50℃下真空干燥。获得呈白色固体的化合物15c(134.0g,83%产率)。m/z 452.21[m+h]
+
。
[0226]
实施例9.化合物15d的合成
[0227][0228]
在25℃至35℃下,向化合物15c(50g,11.1mmol)的meoh/水(1:1,500ml)溶液中缓慢添加naoh(9.8g,24.4mmol)的水(250ml)溶液。将反应混合物搅拌不少于6小时,直到通过uplc确认反应完成。蒸发meoh。通过添加6.0n hcl溶液致使水溶液呈酸性至ph约1-2,并用nacl饱和。用乙酸乙酯(3x750ml)萃取水层。合并乙酸乙酯层,经naso4(100g)干燥并蒸发至干。将粗残余物用己烷(250ml)研磨,过滤,用己烷(100ml)洗涤,并在45℃下真空干燥。获得呈白色固体的化合物15d(20.0g,49%)。m/z 396.11[m+h]
+
。
[0229]
方案4
[0230][0231]
实施例10.化合物13a的合成
[0232][0233]
将化合物15c(80.0g,0.17mol)的thf(2800ml)溶液脱气并用n2吹扫两次。装入10重量%的pd/c(50%湿,8.0g),并将混合物脱气并用h2吹扫两次。将混合物在h2气氛(100psi)下搅拌不少于6小时,直到通过uplc确认反应完成。将混合物脱气并用n2吹扫,经由硅藻土垫过滤。蒸发滤液,将溶剂与乙酸乙酯(2x400ml)交换。在45℃下将残余物溶于乙酸乙酯(400ml)中,并缓慢添加正庚烷(320ml),在45℃下搅拌1h,然后在0至5℃下搅拌1h。
过滤固体,用乙酸乙酯/正庚烷的冷溶液(1:2,160ml)洗涤,并在25至30℃的真空烘箱中干燥,得到呈白色固体的化合物13a(46.2g,82%产率)。m/z 318.14[m+h]
+
。
[0234]
实施例11.化合物13b的合成
[0235][0236]
将dcm和dipea(5.9g,46.4mmol)的溶液冷却至0至5℃。依次装入化合物6(15.0g,23.2mmol)、edc.hcl(5.1g,26.7mmol)、hobt一水合物(4.1g,26.7mmol),同时维持在0至5℃。将混合物搅拌10分钟,装入化合物13a(7.74g,24.3mmol),并在0至5℃下搅拌nlt 21h,直到通过uplc确认反应完成。装入纯化水(150ml),同时维持在低于30℃。分离有机层,用nahco3水溶液(2x105ml)和10%nacl水溶液洗涤。蒸发有机层,将溶剂与乙酸乙酯交换两次(300ml和150ml)。当总体积为4v时,添加正庚烷并在50至55℃加热1h,然后在0至5℃冷却1h。过滤固体,用母液、正庚烷(30ml)洗涤,并在40至45℃下真空干燥固体。获得呈白色固体的化合物13b(17.9g,81%)。m/z 946.05[m+h]
+
。
[0237]
实施例12.化合物13c的合成
[0238][0239]
将化合物13b(10.0g,10.6mmol)的thf(100ml)溶液冷却至0至5℃。在低于5℃下添加koh溶液(1.5g,在50ml水中的26.4mmol)。将反应混合物温热至25至30℃,并搅拌不少于4小时,直到通过uplc确认反应完成。将混合物冷却至0至5℃,用磷酸二氢钠水溶液将ph调节至约7.0,同时将温度维持在低于10℃。装入2-甲基四氢呋喃(150ml)。通过使用1n hcl将ph调节至4-5,同时将温度维持在低于10℃。将混合物在25至30℃下搅拌20分钟。分离有机层,用2-甲基四氢呋喃反洗水层。合并有机层,并用10%nacl水溶液(50ml)洗涤。向有机层中添加三乙胺(4.3g,42.4mmol),搅拌1h,然后蒸发,将溶剂与四氢呋喃(20ml)交换,然后蒸发至完全干燥,并在真空烘箱中进一步干燥。获得呈白色吸湿性固体的化合物13c(tea盐)(9.0g,84%)。m/z 889.13[m+h]
+
。
[0240]
方案5
[0241][0242]
实施例13.化合物12的合成
[0243][0244]
向化合物16-1(41.5g,196.6mmol)的dcm(415ml)溶液中添加n-羟基琥珀酰亚胺(49.7g,432.4mmol)和edc.hcl(82.9g,432.4mmol)。将反应混合物在25℃至30℃下搅拌不少于16小时,直到通过tlc确认形成化合物11b的反应完成。将反应混合物蒸发至2-3v,添加水(415ml),并在25至30℃下将固体搅拌1h。过滤固体,用水(415ml)洗涤,并且在25至30℃下将湿固体用nahco3水溶液(415ml)研磨1h。再次过滤固体,用水(415ml)、mtbe(210ml)洗涤,并在低于45℃下真空干燥,得到呈白色固体的化合物11b(45.0g,56%产率)。
[0245]
将化合物11(110.0g,185.64mmol)的dcm(1100ml)溶液冷却至0至5℃。在低于10℃
下装入化合物11b(33.85g,83.54mmol)和dipea(47.9g,371.3mmol),并在20℃至25℃下搅拌不少于3小时,直到通过uplc确认反应完成。在低于30℃下将水(1100ml)添加到混合物中并搅拌45分钟。分离有机层,用nahco3水溶液(1100ml)、1n hcl(1100ml)和15%nacl水溶液(1100ml)洗涤。蒸发有机层,并用mtbe(500ml)交换溶剂,并溶于mtbe(500ml)中,在25至30℃下搅拌3h。过滤所得固体,用mtbe(250ml)洗涤,并在《50℃下真空干燥。获得呈淡黄色泡沫固体的化合物12(90.0g,86%产率)。m/z 1132.5[m+h]
+
。
[0246]
实施例14.化合物13的合成
[0247][0248]
将化合物12(39g,34.45mmol)的thf(240ml)溶液脱气并用n2吹扫两次。装入10重量%的pd/c(3.9g),并且将混合物脱气并用h2吹扫两次。将反应混合物在h2气氛下搅拌不少于4小时直到通过uplc确认反应完成。将混合物脱气并用n2吹扫,经由硅藻土垫(39g)过滤。蒸发滤液至完全干燥,得到呈灰色泡沫固体的化合物13(36.4g,95%产率)。m/z 1102.5[m+h]
+
。
[0249]
实施例15.化合物14的合成
[0250][0251]
将化合物13(30g,22.72mmol)和n-苄氧羰基甘氨酸(7.97g,38.11mmol)的dcm(150ml)溶液冷却至0至5℃,在低于5℃下在搅拌下依次装入以下物质:tea(7.6ml,54.44mmol)和t3p(29.2ml,49mmol,50%乙酸乙酯溶液)。将反应混合物在0℃至5℃下搅拌不少于3小时直到通过uplc确认反应完成。反应混合物依次用水(110ml)、饱和nahco3水溶液(110ml)、盐水(110ml)洗涤,然后经na2so4(60g)干燥并蒸发至完全干燥,得到呈灰色泡沫固体的化合物14(32g,91%产率)。m/z 1293.6[m+h]
+
。
[0252]
实施例16.化合物15的合成
[0253][0254]
将化合物14(68g,52.78mmol)的thf(400ml)溶液脱气并用n2吹扫两次。装入10重量%的pd/c(6.8g)和tfa(4.4ml,57.84mmol),并且将混合物脱气并用h2吹扫两次。将反应混合物在h2气氛下搅拌不少于4小时直到通过uplc确认反应完成。将混合物脱气并用n2吹扫,经由硅藻土垫(68g)过滤。蒸发滤液至完全干燥,得到呈灰色泡沫固体的化合物15(63g,95%产率)。m/z 1159.6[m+h]
+
(游离碱)。
[0255]
方案6
[0256][0257]
实施例17.化合物16-2的合成
[0258][0259]
在搅拌下,向化合物16-1(50g,236mmol)的dcm(500ml)悬浮液中,依次装入草酰氯(69g,543mmol)和dmf(172mg,2.3mmol),同时将内部温度维持在20至30℃。将混合物在20℃至30℃下搅拌不少于12小时,直到通过uplc由化合物16-1消失确认反应完成。将混合物蒸发至干,并将残余物溶于甲苯。向甲苯溶液中装入t-buoh(52.5g,708mmol)和dmap(66.3g,543mmol)。将此混合物在20℃至25℃下搅拌不少于4小时,直到通过uplc确认反应完成。过滤混合物,滤液用5%柠檬酸水溶液(500ml)、盐水(500ml)洗涤,经naso4(100g)干燥并蒸发。使残余物与正己烷(250ml)共沸,蒸发并干燥,得到呈灰白色固体的化合物16-2(73g,95%产率)。m/z 341.2[m+h]
+
。1h nmr(400mhz,氯仿-d)δ8.92(s,2h),8.87(s,1h),1.64(s,18h)。
[0260]
实施例18.化合物16-3的合成
[0261][0262]
将化合物16-2(30g,92.3mmol)的meoh(360ml)溶液脱气并用n2吹扫两次。装入10重量%的pd/c(3g)并且将混合物脱气并用h2吹扫两次。将混合物在h2气氛下搅拌不少于6小时,直到通过uplc确认反应完成。将混合物脱气并用n2吹扫,经由硅藻土垫过滤。蒸发滤液至完全干燥,得到呈灰白色固体的化合物16-3(26g,95%产率)。m/z 294.2[m+h]
+
。1h nmr(400mhz,氯仿-d)δ7.96(d,j=1.7hz,1h),7.45(d,j=1.5hz,2h),3.92(s,2h),1.59(s,18h)。
[0263]
实施例19.化合物16-4的合成
[0264][0265]
在15至25℃下在搅拌下,向化合物16-3(23.7g,80.7mmol)的dcm(355ml)溶液中依次装入以下物质:n-苄氧羰基甘氨酸(23.7g,113mmol)、tea(16.3g,161mmol)、t3p(92.3g,145mmol,50%乙酸乙酯溶液)。将混合物在15℃至25℃下搅拌不少于2小时,直到通过uplc确认反应完成。混合物依次用水(240ml)、饱和nahco3水溶液(240ml)、盐水(240ml)洗涤,然后经na2so4(48g)干燥,并蒸发至完全干燥,得到呈浅黄色固体的化合物16-4(46.2g,118%产率)。m/z 484.2[m+h]
+
。1h nmr(400mhz,氯仿-d)δ8.95(s,1h),8.34(s,2h),8.29(s,1h),7.36
–
7.27(m,5h),5.99(s,1h),5.16(s,2h),4.12(d,j=5.6hz,2h),1.57(s,18h)。
[0266]
实施例20.化合物16-5的合成
[0267][0268]
将化合物16-4(46.2g,95.3mmol)的meoh(1150ml)溶液脱气并用n2吹扫两次。装入10重量%的pd/c(4.6g),并将混合物脱气并用h2吹扫两次。将混合物在h2气氛下搅拌不少于3小时,直到通过uplc确认反应完成。将混合物脱气并用n2吹扫,经由硅藻土垫过滤。蒸发滤液,将残余物溶于二氯甲烷(500ml)中,蒸发至完全干燥,得到呈浅黄色固体的化合物16-5(32.4g,97%产率)。m/z351.2[m+h]
+
。1h nmr(600mhz,氯仿-d)δ9.63(s,1h),8.37(d,j=1.6hz,2h),8.31(t,j=1.5hz,1h),3.49(d,j=18.7hz,4h),1.59(s,18h)。
[0269]
实施例21.化合物16d的合成
[0270][0271]
向化合物16-5(32.5g,92.7mmol)的二氯甲烷(455ml)溶液中添加化合物15a(11.7g,41.7mmol)和hbtu(52.7g,139mmol)。向此混合物中添加tea(28.1g,278mmol),同时将内部温度维持在15℃至25℃。将反应混合物在相同温度下搅拌不少于6小时,直到通过uplc确认反应完成。反应混合物依次用水(320ml)、nahco3水溶液(320ml)、盐水(320ml)洗涤,经na2so4干燥并蒸发至干。将粗残余物通过柱色谱法纯化(30%至100%ea/己烷),得到呈白色固体的化合物16d(45.2g,51%产率)。m/z 946.5[m+h]
+
。1h nmr(600mhz,dmso-d6)δ8.39(d,j=15.5hz,5h),8.22(t,j=5.7hz,1h),8.05(s,2h),7.35
–
7.24(m,5h),5.06(t,j=9.3hz,2h),4.05(q,j=7.1hz,1h),3.98
–
3.87(m,4h),2.28(hept,j=7.9,7.1hz,2h),1.96(dt,j=18.0,6.7hz,1h),1.82(dd,j=14.5,7.2hz,1h),1.53(d,j=3.5hz,36h)。
[0272]
实施例22.化合物16e的合成
[0273][0274]
向化合物16d(30.0g,31.7mmol)的二氯甲烷(600ml)溶液中添加tfa (108.4g,951mmol),同时将内部温度维持在15℃至25℃。将反应混合物在20℃至25℃下搅拌不少于
12小时,直到通过1h nmr确认反应完成。将混合物蒸发至干,将残余物溶于二氯甲烷(300ml)中,并再次蒸发至干。将所得残余物在二氯甲烷(300ml)与8重量%nahco3水溶液(600ml)之间分配。分离有机层,水层再次用二氯甲烷(300ml)洗涤。丢弃二氯甲烷层。水层用3n hcl(约600ml)酸化,以调节至ph 3-4。过滤形成的固体,用水洗涤,并在45℃下干燥不少于12小时,得到呈白色固体的化合物16e(16.4g,95%产率)。m/z 722.2[m+h]
+
。1h nmr(600mhz,dmso-d6)δ10.31(s,1h),10.11(s,1h),8.48
–
8.38(m,5h),8.30(s,0h),8.25(t,j=5.8hz,1h),8.15(dt,j=3.4,1.6hz,2h),7.67(d,j=6.9hz,1h),7.37
–
7.24(m,5h),5.12
–
5.01(m,2h),4.03(q,j=7.1hz,1h),3.91(dd,j=21.2,6.1hz,4h),2.49(s,0h),2.35
–
2.22(m,2h),1.95(ddt,j=15.0,9.0,5.9hz,1h),1.87
–
1.79(m,1h)。
[0275]
实施例23.化合物16的合成
[0276][0277]
向化合物15(40g,31.82mmol)的dmf(400ml)溶液中添加化合物15a(4.0g,14.32mmol)和hbtu(14.5g,38.18mmol)。将反应混合物冷却至10℃至15℃,添加tea(10.6ml,76.36mmol),同时将内部温度维持在10℃至15℃。将反应混合物温热至20℃至25℃,并搅拌不少于3小时,直到通过uplc确认反应完成。通过添加水(400ml)和乙酸乙酯(400ml)淬灭反应混合物。分离水层,并用dcm(3x400ml)萃取。合并dcm层,用水(5x200ml)洗涤,经na2so4干燥,过滤并蒸发至完全干燥。获得呈淡黄色泡沫固体的化合物16(31.7g,87%产率)。m/z 2563.9[m+h]
+
[0278]
实施例24.化合物16的合成
[0279][0280]
向化合物11(1.0g,1.74mmol)的无水dmf(9ml)溶液中依次添加化合物16e(0.21g,0.29mmol)、edc(333mg,1.74mmol)、hobt(265mg,1.74mmol),并将混合物冷却至0℃至5℃。添加dipea(450mg,3.48mmol),将内部温度维持在0℃至5℃,并在相同温度下搅拌不少于24小时,直到通过uplc确认反应完成。反应混合物用水(11ml)稀释,用二氯甲烷(50ml)萃取。二氯甲烷层用水(2x5ml)洗涤,经na2so4干燥并蒸发至干。将残余物通过柱纯化(2至15%meoh/dcm),得到呈泡沫状固体的纯化合物16(360mg,49.5%产率)。
[0281]
实施例25.化合物16的合成
[0282][0283]
将化合物15d(1.6g,4.08mmol)和化合物13(10g,9.07mmol)的dcm(100ml)溶液冷却至0℃至10℃。在0-10℃下在搅拌下依次装入tea (1.84g,18.14mmol)和t3p(10.34ml,16.3mmol,50%乙酸乙酯溶液)。将反应混合物在25℃至35℃下搅拌不少于6小时,直到通过uplc确认反应完成。通过添加水(200ml)淬灭反应。分离水层,并用dcm(50ml)萃取。合并dcm层,并依次用饱和nahco3水溶液(200ml)、1.0n hcl(200ml)、10%nacl水溶液(200ml)洗涤,然后经na2so4(25g)干燥,并蒸发至约20g。添加mtbe(50ml)并蒸发至完全干燥,并在45℃下进一步真空干燥。获得呈浅黄色固体的化合物16(10.3g,89%产率)。
[0284]
实施例26.化合物17的合成
[0285][0286]
将化合物16(2.7g,1.05mmol)的meoh(27ml)溶液脱气并用n2吹扫两次。装入10重量%的pd/c(0.27g)和tfa(156mg,1.37mmol),并用h2吹扫混合物。将反应混合物在h2气氛下搅拌不少于3小时,直到通过uplc确认反应完成。将混合物脱气并用n2吹扫,经由硅藻土垫过滤。蒸发滤液,将残余物溶于二氯甲烷(25ml)中,蒸发至完全干燥,得到呈灰色泡沫状固体的化合物17(2.4g,90%产率)。m/z 2428.9[m+h]
+
。1h nmr(600mhz,dmso-d6)δ8.54(q,j=5.2hz,1h),8.21(d,j=5.3hz,1h),8.14(t,j=1.8hz,1h),7.95(d,j=8.7hz,1h),7.80(d,j=9.2hz,1h),5.19(d,j=3.4hz,1h),4.95(dd,j=11.2,3.4hz,1h),4.52(d,j=8.5hz,1h),4.07
–
3.97(m,3h),3.94
–
3.82(m,2h),3.76(m,1h),3.59
–
3.47(m,8h),3.44(m,3h),2.38(t,j=7.8hz,1h),2.08(s,3h),1.98(s,3h),1.87(s,3h),1.75(s,3h)。
[0287]
实施例27.化合物18的合成
[0288][0289]
向化合物17(1.0g,0.39mmol)的dcm(25ml)溶液中添加化合物6(0.29g,0.44mmol)、hbtu(186mg,0.49mmol),并将混合物冷却至15℃至25℃。添加dipea(151mg,1.17mmol),将内部温度维持在15℃至25℃,然后在20℃至25℃下搅拌不少于2.5小时,直到通过uplc确认反应完成。反应混合物用dcm(5ml)稀释,用水(10ml)、nahco3水溶液(3x8ml)、盐水(10ml)洗涤,经na2so4干燥并蒸发至干。将残余物通过柱纯化(2至18%meoh/dcm),得到呈灰白色泡沫状固体的纯化合物18(860mg,72.5%产率)。m/z(z=2)1378.5[m-dmtr+2h]
2+
。
[0290]
实施例28.化合物18的合成
[0291][0292][0293]
将化合物13c(39.8g,40.2mmol)和化合物13(93g,84.39mmol)的thf(800ml)溶液冷却至0至10℃。在0-10℃下在搅拌下依次装入tea(0.5ml,3.6mmol)和t3p(20.3g,200.9mmol,50%乙酸乙酯溶液)。将反应混合物在25至35℃下搅拌不少于18小时,直到通过
uplc确认反应完成。通过添加饱和nahco3水溶液(800ml)(10ml)和2-methf(800ml)淬灭反应混合物。分离水层。有机层依次用5%nah2po4(800ml)、10%nacl水溶液(800ml)洗涤,然后经na2so4干燥并蒸发。将溶剂交换成mtbe(400ml),搅拌3-4h并蒸发至完全干燥,并且在45℃下进一步真空干燥。获得呈固体的粗化合物18(120g)。粗物质通过柱色谱法纯化至纯度≥97%并用于下一步骤。m/z(z=2)1378.5[m-dmtr+2h]
2+
。
[0294]
实施例29.化合物19的合成
[0295][0296]
向化合物18(1.1kg,163.6mmol)的dcm(1.1l)溶液中缓慢装入tea(126g,1260mmol),将温度保持在25℃。然后分批装入化合物18a(126g,1260mmol),将温度维持在25℃。将所得混合物在40至45℃下搅拌不少于72小时,直到通过uplc确认反应完成。将反应混合物冷却至20℃至25℃,并用nahco3水溶液(2x5l)洗涤,经na2so4干燥,过滤并蒸发至完全干燥。获得呈灰白色固体的化合物19(1.0kg)。对于游离酸:m/z(z=2)1428.5[m-dmtr+2h]
2+
。
[0297]
实施例30.式13cc化合物的结晶钾盐的合成:
[0298][0299]
将5.0g(1.0当量)的化合物13bb和25ml(5v)的meoh装入反应器(100ml)中。溶解后,将内容物调节至0-5℃。在另一个反应器中,将653mg(2.2当量)koh用25ml(5v)的meoh溶解。将meoh中的koh缓慢装入到内容物中,并将反应混合物缓慢调节至40℃。搅拌反应混合物直到反应完成。浓缩至最小体积后,装入10v的cpme,并在50-60℃下搅拌内容物。在搅拌期间,形成浅黄色浆液。在减压下将浆液浓缩至最小体积。装入10v庚烷后,将浆液在50-60
℃下搅拌1小时,并缓慢调节至0-5℃。搅拌1小时后,将内容物经由滤纸过滤,并用2v的庚烷洗涤湿饼。获得5.2g呈灰白色固体的产物。
[0300]
所有公开、专利和专利文件均以引用方式并入本文,如同以引用方式将其单独并入一样。已经参考各种具体和优选的实施方案和技术描述了本发明。然而,应当理解,在不脱离本发明的精神和范围的情况下,可进行许多变化和修改。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1