一种吡啶酮修饰锌酞菁及其制备方法和应用
1.本发明属于抗肿瘤光敏剂,具体涉及一种吡啶酮修饰锌酞菁及其制备方法和应用。
背景技术:
2.恶性肿瘤是威胁人类健康的重大疾病之一。光动力疗法(photodynamic therapy,简称pdt)是一种已被批准用于临床的肿瘤治疗方法。光动力疗法的原理是,光照富集于肿瘤组织的光敏剂,光敏剂被激发后,与周围的氧气反应生成活性氧,杀伤肿瘤细胞。激发光敏剂的光源穿透深度不足,光敏剂产生单线态氧的利用率低,以及乏氧条件下缺氧诱导因子
‑
1(hypoxia inducible factor
‑
1,hif
‑
1)上调对治疗产生抗性是pdt治疗过程中三大瓶颈问题。
3.传统光敏剂的激发波长多在可见光范围内。可见光组织穿透深度不足,因此,目前临床上pdt多用于浅表肿瘤治疗。近红外(near infrared,nir)光的组织穿透能力强且在生物组织中的传输效率高,非常适用于深部肿瘤的pdt治疗。因此,可见光及近红外光均可激发的光敏剂可实现浅表和深部肿瘤的同步治疗。
4.单线态氧(singlet oxygen,1o2)是pdt中首要的活性氧物种,具有很强的氧化损伤肿瘤细胞的能力。但是,其在生理环境中的寿命非常短(<40μs)。因此,其在pdt中的利用率有限,许多1o2可能在氧化损伤肿瘤细胞之前即发生淬灭损失。
5.此外,乏氧微环境是实体肿瘤微的重要特征。且pdt治疗本身还是一个耗氧过程。pdt治疗进一步加重肿瘤缺氧。在乏氧条件下,hif
‑
1表达上调。hif
‑
1上调会促进肿瘤组织血管生成,触发肿瘤转移和细胞对凋亡的抗性。此外,hif
‑
1上调可以增强细胞对包括pdt在内的各种癌症治疗的抵抗力。因此,hif
‑
1表达水平与患者死亡率和治疗耐受性呈正相关。
技术实现要素:
6.发明目的:针对pdt治疗过程中存在的问题,本发明提供了本发明提供一种新型吡啶酮修饰锌酞菁,该新型锌酞菁被肿瘤细胞摄取后,可降低缺氧诱导因子
‑
1的表达水平,可被红及近红外光激发产生活性氧,可储存多余的单线态氧并在生理温度下缓慢释放,最终获得良好的光动力抗肿瘤活性,该新型酞菁适合光动力疗法光敏抗肿瘤治疗。
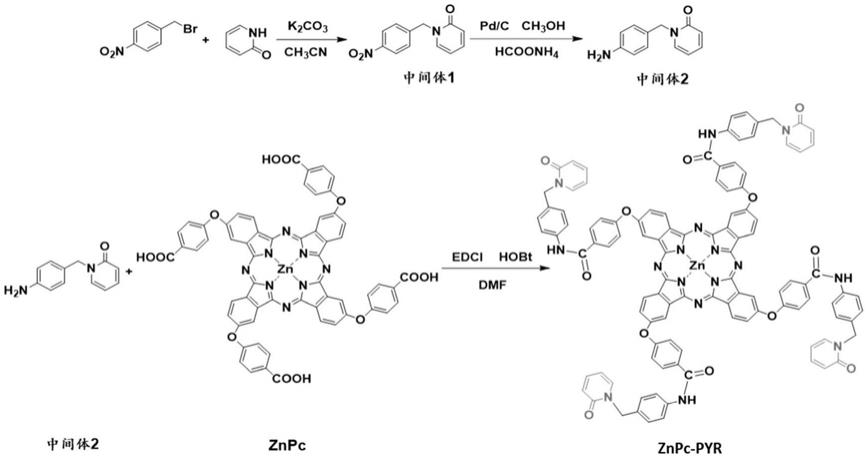
7.本发明还提供所述吡啶酮修饰锌酞菁的制备方法及其应用。
8.技术方案:为了实现上述目的,本发明所述一种吡啶酮修饰锌酞菁,所述吡啶酮修饰锌酞菁的结构式如下所示:
[0009][0010]
本发明所述的吡啶酮修饰锌酞菁的制备方法,包括如下步骤:
[0011]
(1)中间体1合成:在ch3cn中,将4
‑
硝基溴化苄,2
‑
羟基吡啶和k2co3混合,在惰性气体气氛下搅拌过夜,产物减压干燥后与水混合,萃取干燥,纯化后得到中间体1;
[0012]
(2)中间体2的合成:在甲醇中,将中间体1、hcoonh4、pd/c混合,在惰性气体气氛下于反应,产物减压干燥后溶解萃取干燥,然后过滤干燥,得到中间体2;
[0013]
(3)吡啶酮修饰锌酞菁znpc
‑
pyr的合成:在dmf中,将znpc、1
‑
乙基
‑
(3
‑
二甲基氨基丙基)碳二亚胺盐酸盐(edc
·
hcl)、1
‑
羟基苯并三唑一水合物(hobt)混合,将混合物搅拌将溶解于dmf中的中间体2滴入上述混合液,继续搅拌1然后将上述溶液倒入丙酮通过离心收集沉淀物,将沉淀物溶于酸性水溶液中,离心后得到上清液,调节ph再次通过离心纯化固体产物,冲洗真空干燥后,得到吡啶酮修饰锌酞菁znpc
‑
pyr;
[0014]
其反应式如下所示:
[0015][0016]
其中,步骤(1)所述4
‑
硝基溴化苄,2
‑
羟基吡啶和k2co3的摩尔比为1:1~2:1~2;ch3cn的用量为10~50ml;k2co3的用量为1.45~4.35g;反应温度为50℃~100℃。反应时间为2h~24h。
[0017]
作为优选,所步骤(1)所述反应温度为80℃在氮气气氛下搅拌过夜。
[0018]
其中,步骤(2)所述中间体1的用量为0.1~2g,hcoonh4的用量为0.1~3g,pd/c的
用量为0.02~0.3g;反应温度为0℃~80℃;反应时间为0.5h~24h。
[0019]
作为优选,所述步骤(2)在氮气气氛下于室温下反应6小时。
[0020]
其中,步骤(3)所述中间体2、znpc、edc
·
hcl及hobt的摩尔比为8~10:1:8~10:8~10;其中两次dmf用量第一次1
‑
10ml,第二次1
‑
5ml。混合物反应温度为0℃~60℃。反应时间为0.5h~24h。
[0021]
作为优选,所述步骤(2)将混合物在25℃下搅拌1小时,将溶解于dmf中的中间体2滴入上述混合液,在0℃下继续搅拌1h。
[0022]
本发明中使用的znpc的合成路线如下:
[0023][0024]
具体合成过程可参考文献:photosensitizer and autophagy promoter coloaded ros responsive dendrimer
‑
assembled carrier for synergistic enhancement of tumor growth suppression,small 2018,14,1802337
[0025]
本发明所述的吡啶酮修饰锌酞菁在制备光动力药物中的应用。
[0026]
其中,所述吡啶酮修饰锌酞菁在制备光动力药物中作为光敏剂,具有良好的光动力抗肿瘤活性,适合光动力疗法光敏抗肿瘤治疗。
[0027]
本发明的联吡啶修饰锌酞菁具有多重抗肿瘤机制,被肿瘤细胞摄取后,可降低缺氧诱导因子
‑
1的表达水平,降低肿瘤细胞对pdt治疗的耐受性;可被红(665nm)及近红外光(808nm)激发产生活性氧,实现浅表及深部肿瘤的同步治疗;吡啶酮结构可储存多余的1o2,并在生理温度(37℃)下缓慢释放,提升1o2的利用效率。最终获得良好的光动力抗肿瘤活性。
[0028]
传统的锌酞菁光敏剂是由红光激发的,红光对组织的穿透深度有限,只适合治疗浅表肿瘤。相比之下,近红外(700
‑
1000nm)比红光有更大的穿透深度,更适合治疗大而深的肿瘤。本发明的znpc
‑
pyr在665nm左右具有较强的吸收强度,说明它可以被红光激发产生ros用于浅表肿瘤的治疗。此外,znpc
‑
pyr在800nm左右也有尾部吸光度,在近红外光可以激发产生ros用于深部肿瘤的治疗。
[0029]
有益效果:与现有技术相比,本发明具有如下优点:
[0030]
(1)本发明的吡啶酮修饰锌酞菁znpc
‑
pyr被肿瘤细胞摄取后,可降低缺氧诱导因子
‑
1的表达水平,降低肿瘤细胞对pdt治疗的耐受性;
[0031]
(2)本发明所制得的znpc
‑
pyr可被红(665nm)及近红外光(808nm)激发产生活性氧,实现浅表及深部肿瘤的同步治疗;
[0032]
(3)本发明所制得的znpc
‑
pyr可储存1o2,并在生理温度(37℃)下缓慢释放,提升1o2的利用效率;
[0033]
(4)本发明的znpc
‑
pyr的制备方法简单方便,原料来源广,活性高,增敏机制多样化,可作为光敏剂应用在制备光动力药物中,具有良好的光动力抗肿瘤活性。
附图说明
[0034]
图1为本发明的吡啶酮修饰锌酞菁znpc
‑
pyr的合成流程图;
[0035]
图2为znpc
‑
pyr降低细胞内hif
‑
1α表达水平示意图;
[0036]
图3为hela细胞在常氧(a1)和乏氧(a2)条件下,不同光照条件下的细胞活性图;4t1细胞在常氧(b1)和乏氧(b2)条件下,不同光照条件下的细胞活性图;
[0037]
图4为znpc
‑
pyr在665nm及808nm光激发条件下,产生单线态氧示意图;
[0038]
图5为不同光照条件下znpc
‑
pyr的体外ros生成示意图;
[0039]
图6为znpc
‑
pyr储存释放单线态氧示意图;
[0040]
图7为665nm和808nm光照后,znpc
‑
pyr在细胞内1o2的产生、储存与释放示意图;
[0041]
图8为皮下移植瘤模型小鼠内,znpc
‑
pyr在665nm、808nm及665nm+808nm光激发条件下抑制肿瘤生长的能力比较示意图。
具体实施方式
[0042]
根据下述实施例,可以更好地理解本发明。本领域的技术人员容易理解,实施例所描述的内容仅用于说明本发明,而不应当也不会限制权利要求书中所详细描述的本发明。下述实施例中所使用的材料、试剂等,如无特殊说明,均可从商业途径得到。实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂家建议的条件。
[0043]
实施例1
[0044]
(1)在20ml的ch3cn中,加入4
‑
硝基溴化苄(2.3g),2
‑
羟基吡啶(1g)和k2co3(2.9g)混合,在氮气气氛下于80℃搅拌过夜。反应完成后,产物减压干燥后与水混合,用dcm萃取,无水na2so4干燥,柱层析(pe/ea=5/1,dcm/meoh=100/1,v/v)纯化得到中间体1;
[0045]
制备的中间体1化合物:1h nmr(400mhz,d6‑
dmso),δ(ppm)8.22(d,j=8.8hz,2h),7.86(dd,j
12
=2.0hz,j
23
=6.8hz,1h),7.53
‑
7.44(m,3h),6.45(d,j=8.8hz,1h),6.30(td,j
12
=1.6hz,j
23
=5.2hz,1h),5.24(s,2h).
13
c nmr(100mhz,d6‑
dmso):δ(ppm)161.88,147.27,145.54,140.96,139.73,129.06,124.16,123.86,120.45,106.29,51.41.ms
‑
esi(m/z):calculated for c
12
h
10
n2o
3 230.07.found[m
‑
h]
‑
229.0.
[0046]
(2)在20ml的meoh中,加入中间体1(1g,4.3mmol)、hcoonh4(1.5g)和pd/c(150mg,10%pd)混合,并在氮气气氛下,于室温下反应6小时。将pd/c过滤后产物减压干燥后再溶解在dcm中,用na2co3溶液洗涤3次,并将水相用dcm萃取。合并的萃取物用无水na2so4干燥,过滤后旋蒸去除溶剂,得到中间体2;
[0047]
制备的中间体2化合物:1h nmr(400mhz,d6‑
dmso),δ(ppm)7.69(dd,j
12
=2.4hz,j
23
=6.8hz,1h),7.41
‑
7.34(m,1h),7.02(d,j=8.4hz,2h),6.50(d,j=8.4hz,2h),6.38(dd,j
12
=0.8hz,j
23
=8.4hz,1h),6.19(td,j
12
=1.2hz,j
23
=5.6hz,1h),5.22
‑
5.00(m,2h),4.88(s,2h).
13
c nmr(100mhz,d6‑
dmso):δ(ppm)161.83,148.65,140.15,139.23,129.68,124.70,120.18,114.22,105.76,51.09.ms
‑
esi(m/z):calculated for c
12
h
10
n2o
3 200.09.found[m
‑
h]
‑
201.2.
[0048]
3)在5ml的dmf中,将znpc(56mg)、edc
·
hcl(76.68mg)和hobt(54.05mg)混合,在25℃下搅拌1小时。上述混合物置于冰水浴中,将溶解于2mldmf的中间体2(80mg)逐滴加入上述混合物。在0℃下搅拌反应1小时后,然后将混合物倒入丙酮(80ml)中,通过离心收集沉淀
物,将其溶于酸性水溶液(ph=4)中,离心后得到上清液,并用10%(wt%)的naoh将上清液ph调节至10,再次通过离心纯化固体产物,然后用ea冲洗数次,真空干燥后得到吡啶酮修饰酞菁衍生物znpc
‑
pyr。制备吡啶酮修饰酞菁衍生物的流程图如图1所示。
[0049]
制备的znpc
‑
pyr:1h nmr(400mhz,d6‑
dmso):δ(ppm)10.32(dd,j
12
=6.4hz,j
23
=25.6hz,4h),9.04
‑
8.36(m,12h),8.20(d,j=34.4hz,8h),7.79(s,16h),7.47
‑
7.20(m,16h),6.44(d,j=8.8hz,4h),6.25(s,4h),5.08(s,8h).
13
c nmr(100mhz,d6‑
dmso):δ(ppm)165.14,162.79,161.90,161.75,160.52,160.33,157.35,140.49,139.65,139.47,139.10,133.77,133.02,133.60,128.68,124.34,121.01,120.33,118.92,118.54,106.01,51.19.
[0050]
实施例2
[0051]
实施例2与实施例1的制备方法相同,不同之处在于:步骤(1)中4
‑
硝基溴化苄与2
‑
羟基吡啶的摩尔比为1:2,2
‑
羟基吡啶(0.5g),ch3cn的用量为10ml;k2co3的用量为1.45g;反应温度为50℃,反应时间为24h;步骤(2)中间体1的用量为0.1g,hcoonh4的用量为0.1g,pd/c的用量为0.02g;反应温度为0℃;反应时间为24h;步骤(3)中间体2、znpc、edc
·
hcl及hobt的摩尔比为10:1:10:10;其中两次dmf用量均为1ml,混合物反应温度为0℃。反应时间为24h。
[0052]
实施例3
[0053]
实施例3与实施例1的制备方法相同,不同之处在于:步骤(1)中4
‑
硝基溴化苄与2
‑
羟基吡啶的摩尔比为1:1,2
‑
羟基吡啶(1.5g),ch3cn的用量为50ml;k2co3的用量为4.35g;反应温度为100℃,反应时间为2h;步骤(2)中间体1的用量为2g,hcoonh4的用量为3g,pd/c的用量为0.3g;反应温度为80℃;反应时间为0.5h;步骤(3)中间体2、znpc、edc
·
hcl及hobt的摩尔比为8:1:10:10;其中两次dmf用量第一次10ml,第二次5ml,混合物反应温度为60℃。反应时间为0.5h。
[0054]
实施例4
[0055]
znpc
‑
pyr降低细胞内hif
‑
1α表达水平检测
[0056]
znpc
‑
pyr的水溶性低。因此,在dmso中制备了znpc
‑
pyr的储备溶液浓度为10
‑3mol/l。添加到水溶液中后,znpc
‑
pyr趋于聚集沉淀。cremophor el是一种具有优异溶解性和乳化活性的非离子表面活性剂。而且cremophor el毒性极低,被广泛用于提高药物溶解度。因此,在本发明中,将0.001%的cremophor el用作znpc
‑
pyr的增溶剂用于所有理化性质及活性研究。
[0057]
缺氧诱导因子1(hif
‑
1)由组成性表达的亚基(hif
‑
1β)和o2调节的亚基(hif
‑
1α)组成。hif
‑
1α的下调可以代表hif
‑
1的表达情况。hela细胞培养于缺氧培养箱(1%o2、5%co2、37℃)中进行缺氧预处理。培养24小时后,将细胞使用含实施例1所获得的znpc
‑
pyr的1ml的无血清dmem培养基([znpc
‑
pyr]=6μm)处理4小时,使用含有edta(5mm)的细胞裂解液裂解细胞提取蛋白,edta可作为铁螯合剂降低细胞内铁离子的浓度抑制hif
‑
1α的快速降解。使用sds
‑
page电泳及ecl成像技术检测znpc
‑
pyr降低细胞内hif
‑
1α表达水平效果。设置空白组、dmso组及cremophor el组作为对照证明细胞中hif
‑
1α下调源自znpc
‑
pyr。
[0058]
如图2所示,dmso组及cremophor el组并未引起细胞hif
‑
1α表达水平下调。但znpc
‑
pyr可显著下调肿瘤细胞hif
‑
1α表达水平,hif
‑
1α的上调在血管生成、转移和凋亡抵抗等肿瘤进展过程中起主导作用,hif
‑
1α的上调与pdt治疗耐药相关。因此,本发明的znpc
‑
pyr可以抑制hif
‑
1α是提高pdt治疗效率的有效途径。
[0059]
细胞均培养在三气培养箱(1%o2,5%co2,37℃)中,乏氧实验所用到的溶液均用氮气除氧处理。细胞在96孔板中培养24小时,除去原先培养液,加入含有znpc
‑
pyr(6μm)的新鲜培养基,在培养箱中孵育4小时,665nm(1w cm
‑2)的激光器照射4min,808nm(1w cm
‑2)的激光器照射4min,继续在培养箱孵育过夜。每孔加入100μl10%mtt的新鲜培养基,4小时后用dmso溶解甲瓒结晶,利用酶标仪测定570nm处的光密度od值,甲瓒结晶的生成量与活细胞数目成。如图3所示,在低氧条件下,znpc
‑
pyr对hela细胞也具有良好的双光诱导pdt活性。在4t1细胞上也进行了相同的实验,得到了相似的结果。由于实体肿瘤中同时存在缺氧区和常氧区,因此这一特性对pdt的体内治疗具有重要意义。(图3中a1、b1是在常氧条件下hela和4t1细胞的毒性;a2、b2是在常氧条件下hela和4t1细胞的毒性),说明znpc
‑
pyr的hif
‑
1下调功能可以增强肿瘤细胞对缺氧条件下pdt治疗的敏感性。
[0060]
实施例5
[0061]
znpc
‑
pyr在665nm及808nm光激发条件下,产生单线态氧检测
[0062]
adpa与1o2反应生成其内过氧化物,进而导致adpa特征吸收峰处(λmax=378nm)吸光强度的降低。通过监测adpa在水溶液中被氧化而引起的吸收强度下降,来研究1o2的产生。
[0063]
使用了商用单重态氧探针adpa(9,10
‑
二苯基蒽丙酸,西格玛奥德里奇(上海)有限公司)来检测该过程,单线态氧可将adpa氧化为无吸收的内过氧化物,导致其吸收值下降。将实施例1制备的znpc
‑
pyr溶解在0.001%crel的水溶液中([znpc
‑
pyr]=10μm),然后与adpa(100μm)混合(znpc
‑
pyr溶液与adpa水溶液体积比10:1)。665nm(0.4w cm
‑2)led及808nm(0.25w cm
‑2)激光器用作激发光源。记录光照时间后,混合溶液的吸收光谱。
[0064]
如图4所示,在665nm及808nm光照射的条件下,随着光找时间的延长,adpa吸收光谱强度均逐渐下降。说明znpc
‑
pyr即可被665nm激光激发,也可被808nm激光激发产生单线态氧
[0065]
以dcfh
‑
da为总活性氧探针(西格玛奥德里奇(上海)有限公司),测试znpc
‑
pyr体外总ros生成能力。细胞内ros可氧化无荧光的dcfh产生有荧光的dcf,绿色荧光强度与细胞内ros水平成正比。具体过程:将hela细胞在含10%nbs的dmem中培养,置于37℃、5%co2的加湿培养箱中。在激光共聚焦培养皿中培养过夜后,与znpc
‑
pyr(6μm)共孵育4小时。之后,弃去原来培养液,将hela细胞与dcfh
‑
da荧光探针共孵育一小时,665nm(0.4w cm
‑2)光照3分钟,808nm(0.25w cm
‑2)光照15分钟,665+808nm(665nm led光照3分钟后808nm led光照15分钟)。收集细胞后,用流式细胞仪定量检测488nm激发时的绿色荧光强度。在最大激发波长488nm和最大发射波长525nm,用激光共聚焦观察活细胞的绿色荧光信号。
[0066]
如下图5所示,在665和808nm光照下,znpc
‑
pyr能在癌细胞内有效生成ros。此外,665+808nm光照射的细胞总ros水平显著高于单独665或808nm处理的细胞。说明znpc
‑
pyr可以在665nm和808nm的双波长激发下产生大量ros来氧化损伤肿瘤细胞,能够实现浅表和深部肿瘤的pdt治疗。
[0067]
实施例6
[0068]
znpc
‑
pyr储存释放单线态氧检测
[0069]
为了验证在非光照条件下连续释放单重态氧的znpc
‑
pyr的可行性,使用了商用单重态氧探针adpa来检测该过程。在运行检测之前,将实施例1制备的znpc
‑
pyr([znpc
‑
pyr]
=10μm)溶解在0.001%crel的水溶液中,然后与adpa(100μm)混合(znpc
‑
pyr溶液与adpa水溶液体积比10:1)。665nm(0.4w cm
‑2)led被用作激发光源。记录间断光照及光照间隙的加热过程(37℃、黑暗)中adpa的吸收光谱(光照射30s后,避开光30分钟,该循环进行4次)。
[0070]
如图6所示,光照过程可导致adpa在378nm(λ
max of adpa)吸收强度下降,说明光照可触发znpc
‑
pyr发生pdt过程产生单线态氧;同样的,暗处存放时,生理温度仍可触发adpa在378nm吸收强度继续下降。说明znpc
‑
pyr的吡啶酮结构可以储存znpc
‑
pyr的pdt过程产生的单线态氧,并在生理温度下,缓慢释放储存的单线态氧,该过程可以提升1o2的利用效率,进而提升pdt治疗效果。
[0071]
此外,通过hela细胞内1o2生成检测进一步证实了znpc
‑
pyr在665和808nm光照下的1o2存储和释放功能。以sosg为探针(赛默飞世尔科技(中国)有限公司),采用激光共聚焦和细胞流式仪检测细胞内1o2生成。具体过程:使用细胞流式仪和激光共聚焦来检测药物在细胞内具有的1o2储存与释放能力。hela细胞在6孔板或激光共聚焦培养皿中培养过夜,znpc
‑
pyr(6μm)孵育4小时,弃去原先培养液加入无血清的新鲜dmem,一组立即光照,放入培养箱继续孵育,6小时后另一组光照,两组同时加入sosg单线态氧探针(5μm),培养箱孵育一个小时后,收集细胞用细胞流式仪检测荧光强度或用pbs清洗三遍,用激光共聚焦观察荧光强度。665nm led灯照射3分钟,808nm led灯照射15分钟。
[0072]
如下图7所示,znpc
‑
pyr在665或808nm光照下,癌细胞立即产生大量的1o2。两组光照后的细胞在37℃的培养箱中黑暗中连续孵育6h后,细胞荧光强度继续增强,说明znpc
‑
pyr细胞内可以连续释放1o2。
[0073]
实施例7
[0074]
体内抗癌活性检测
[0075]
小鼠皮下移植瘤模型动物体内,znpc
‑
pyr在不同光照条件下抑制肿瘤生长的能力比较实验。实验步骤如下,将znpc
‑
pyr通过尾静脉注射到荷瘤裸鼠体内,6h后(给药浓度为5mg/kg小鼠),使用不同光照条件光照肿瘤位置(665nm组:665nm led功率为0.9w/cm2,光照时间为30分钟;808nm组:808nm激光器功率为0.3w/cm2,光照时间为7分钟;665nm+808nm组:665nm led功率为0.9w/cm2,光照时间为30分钟,808nm激光器功率为0.3w/cm2,光照时间为7分钟)。在第0,2,4,6,8,10,12天给药,从第一天开始,每天进行光照。整个治疗过程持续14天,每天测量肿瘤组织尺寸,记录瘤体积变化规律。
[0076]
如图8所示,体内抗肿瘤活性研究结果显示,665nm+808nm组的抑制肿瘤生长的能力显著强于665nm组及808nm组。说明znpc
‑
pyr可被665nm和808nm激发产生活性氧。比665nm相比,808nm的光具有更好的中深部组织穿透能力。因此,该znpc
‑
pyr可实现深表与浅表肿瘤高效pdt治疗。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1