一种用于原发性肝细胞癌早期诊断的生物标志物及其检测试剂和应用的制作方法
no:44所示的gut metagenome phascolarctobacterium sp.、具有16s rrna核苷酸序列如seq id no:45所示的uncultured bacterium phascolarctobacterium sp.、具有16s rrna核苷酸序列如seq id no:46所示的phascolarctobacterium faecium;所述巨单胞菌属包括具有16s rrna核苷酸序列如seq id no:47所示的uncultured bacterium megamonas sp.、具有16s rrna核苷酸序列如seq id no:48所示的megamonas funiformis。
11.本发明提供了一种检测所述生物标志物的试剂在制备筛查和诊断肝癌早期的试剂盒中的应用。
12.本发明提供了一种用于检测所述生物标志物的检测试剂,所述检测试剂包括用于扩增16s rrna的引物对;所述引物对包括上游引物和下游引物;所述上游引物的核苷酸序列如seq id no:51所示;所述下游引物的核苷酸序列如seq id no:52所示;其中n=a、g、c或t,w=a或t,h=a、c或t,v=a、g或c。
13.本发明提供了一种用于诊断肝癌早期的试剂盒,包括所述检测试剂。
14.本发明提供了一种用于肝癌早期诊断的预测模型的构建方法,包括以下步骤:
15.1)分别从健康人群和肝癌早期患者的粪便中提取细菌dna,得到健康人群组的细菌dna和肝癌早期患者组的细菌dna;
16.2)对所述健康人群组的细菌dna和肝癌早期患者组的细菌dna分别进行16s dna扩增、文库构建和16s rrna测序,对测序数据进行生物信息学分析,得到两组样本中微生物属水平间的丰度差异;
17.3)根据所述两组样本中微生物属水平间的丰度差异筛选有显著差异的生物标志物;
18.4)通过所述生物标志物建立特异性鉴定肝癌早期和健康人群样本的随机森林模型,评估,得到肝癌早期诊断的预测模型。
19.优选的,步骤2)中所述生物信息学分析的方法为通过dada2对测序数据进行过滤、去噪、拼接、去嵌合体形成16s rrna基因的序列变异体,参考silva数据库对所述序列变异体进行分类注释,并通过stamp中welch
‘
s t
‑
test检验分析肝癌早期患者和健康人群中肠道微生物属水平间的丰度差异。
20.优选的,筛选有显著差异的生物标志物的方法为用线性判别分析和效应大小方法进行筛选。
21.优选的,步骤4)中通过所述生物标志物建立特异性鉴定肝癌早期和健康人群样本的随机森林模型的方法为将全部肝癌早期样本和健康人群样本分别按数量比4:1分为训练集和隔离集,使用python软件sklearn.ensemble.randomforestclassifier模块对所述训练集进行随机森林模型训练;
22.所述评估的方法包括通过绘制学习曲线评估模型的拟合情况,同时通过接受者操作特性曲线评估模型效果,所述隔离集用于最终模型准确性评估。
23.优选的,所述随机森林模型训练包括使用网格搜索方法调整模型训练参数,用十次交叉验证训练随机森林模型;
24.所述模型训练参数设定如下:梯度n_estimators=[100,500,1000];max_depth=[1,2,3,7,9];max_features=["log2","sqrt"];
[0025]
所述十次交叉验证训练随机森林模型的方法为使用python软件sklearn.model_
selection.gridsearchcv模块搜索最佳参数,共计30种参数组合,每种参数组合下将训练集分割成10份子样本,一份单独的子样本被保留作为验证模型的数据,其他9份子样本用来训练;重复10次,每份子样本验证一次,平均10次的结果作为该参数组合下最终模型结果;总计模型训练次数为300次。
[0026]
本发明提供的用于原发性肝细胞癌早期诊断的生物标志物,包括以下菌属:布劳特氏菌属、大肠埃希菌
‑
志贺氏菌属、胃球菌属、链球菌属、真杆菌属、多尔氏菌属、拟杆菌属、普雷沃氏菌属9、毛螺菌属、粪杆菌属、考拉杆菌属、巨单胞菌属、lachnospiraceae nk4a136 group、梭状芽胞杆菌属,共14个属。本发明利用16s rrna对肠道微生物进行测序,鉴定出肝癌早期特异性的生物标志物,通过检测生物标志物的丰度上调或下调的变化,实现患者肝癌早期的风险评估,本发明将上述14属的特征微生物作为生物标志物进行诊断,具有较高的精度、召回率和特异度,分别为0.97、1和0.98。
[0027]
本发明提供的用于检测所述生物标志物的检测试剂,包括用于扩增16s rrna的引物对。所述引物对为上述14个微生物菌属的16s rrna的通用扩增引物,能够同时实现对14个属的几十种细菌实现特异性扩增,基于生物标志物指示样本中患肝癌早期的风险的功能,通过检测生物标志物实现诊断样本是否患肝癌早期风险的目的。同时本发明采用一对引物进行pcr扩增,具有检测简便,技术要求低,诊断成本低等优点;本发明的检测对象为供试者的粪便,简单方便,并且无创性更易让患者接受。
附图说明
[0028]
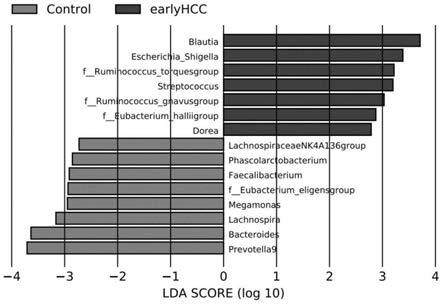
图1为通过线性判别分析效应大小鉴定肝癌早期特异性的肠道生物标志物;
[0029]
图2为肝癌早期患者和健康对照生物标志物的丰度差异;
[0030]
图3为基于肠道生物标志物建立随机森林模型的学习曲线;
[0031]
图4为基于肠道生物标记物对肝癌早期患者和健康对照的诊断能力。
具体实施方式
[0032]
本发明提供了一种用于原发性肝细胞癌早期诊断的生物标志物,包括以下菌属:布劳特氏菌属、大肠埃希菌
‑
志贺氏菌属、胃球菌属、链球菌属、真杆菌属、多尔氏菌属、拟杆菌属、普雷沃氏菌属9、毛螺菌属、粪杆菌属、考拉杆菌属、巨单胞菌属、lachnospiraceae nk4a136 group。
[0033]
在本发明中,从141例肝癌早期患者粪便样本和194例健康人群粪便样本中经过提取dna、16s rrna扩增及测序,生物信息学分析,显著性差异分析,得到在肝癌早期患者和健康人群间丰度变化显著的特定菌属微生物即为生物标志物(见图1)。所述布劳特氏菌属包括具有16s rrna核苷酸序列如seq id no:3所示的blautia hydrogenotrophica、具有16s rrna核苷酸序列如seq id no:4所示的blautia sp.、具有16s rrna核苷酸序列如seq id no:5所示的blautia faecis、具有16s rrna核苷酸序列如seq id no:6所示的uncultured blautia sp.、具有16s rrna核酸序列如seq id no:7所示的uncultured bacterium blautia sp.、具有16s rrna核苷酸序列如seq id no:8所示的blautia obeum、具有16s rrna核苷酸序列如seq id no:9所示的blautia wexlerae;所述大肠埃希菌
‑
志贺氏菌属包括具有16s rrna核苷酸序列如seq id no:1所示的escherichia coli、具有16s rrna核苷
phascolarctobacterium sp.、具有16s rrna核苷酸序列如seq id no:45所示的uncultured bacterium phascolarctobacterium sp.、具有16s rrna核苷酸序列如seq id no:46所示的phascolarctobacterium faecium;所述巨单胞菌属包括具有16s rrna核苷酸序列如seq id no:47所示的uncultured bacterium megamonas sp.、具有16s rrna核苷酸序列如seq id no:48所示的megamonas funiformis。
[0036]
在本发明中,布劳特氏菌属、大肠埃希菌
‑
志贺氏菌属、链球菌属、胃球菌属(包括扭链瘤胃球菌ruminococcus gnavus group和活泼瘤胃球菌ruminococcus torques group)、真杆菌属的霍氏真杆菌(eubacterium hallii group)、多尔氏菌属在肝癌早期人群的粪便中相对丰度显著升高;而拟杆菌属、普雷沃氏菌属9、毛螺菌属、真杆菌属的挑剔真杆菌(eubacterium eligens group)、粪杆菌属、考拉杆菌属、巨单胞菌属、lachnospiraceae nk4a136 group在肝癌早期人群的粪便中相对丰度显著降低,菌属的丰度见表1。
[0037]
表1标志物菌属的相对丰度表
[0038][0039][0040]
本发明提供了一种检测所述生物标志物的检测试剂在制备筛查和诊断肝癌早期的试剂盒中的应用。在本发明中,所述检测试剂包括用于特异性扩增所述生物标志物的引物。所述引物为用于扩增16s rrna的引物对;所述引物对包括上游引物和下游引物;所述上游引物的核苷酸序列如seq id no:51所示(cctacgggnggcwgcag);所述下游引物的核苷酸序列如seq id no:52所示(gactachvgggtatctaatcc);其中n=a、g、c或t,w=a或t,h=a、c或t,v=a、g或c。本发明对所述引物的来源不做具体限定,采用本领域所熟知的引物扩增来源即可。在本发明实施例中,所述引物委托北京擎科生物科技有限公司合成。
[0041]
本发明提供了一种用于诊断肝癌早期的试剂盒,包括所述检测试剂,还优选包括pcr反应用混合液。本发明对所述pcr反应用混合液的来源不做具体限定,采用本领域所熟知的pcr反应用混合液即可。在本发明实施例中,所述pcr反应用混合液购自北京全式金生物技术(transgen biotech)有限公司。
[0042]
本发明还提供了一种用于肝癌早期诊断的预测模型的构建方法,包括以下步骤:
[0043]
1)分别从健康人群和肝癌早期患者的粪便中提取细菌dna,得到健康人群组的细菌dna和肝癌早期患者组的细菌dna;
[0044]
2)对所述健康人群组的细菌dna和肝癌早期患者组的细菌dna分别进行16s dna扩增、文库构建和16s rrna测序,对测序数据进行生物信息学分析,得到两组样本中微生物属水平间的丰度差异;
[0045]
3)根据所述两组样本中微生物属水平间的丰度差异筛选有显著差异的生物标志物;
[0046]
4)通过所述生物标志物建立特异性鉴定肝癌早期和健康人群样本的随机森林模型,评估,得到肝癌早期诊断的预测模型。
[0047]
本发明分别从健康人群和肝癌早期患者的粪便中提取细菌dna,得到健康人群组的细菌dna和肝癌早期患者组的细菌dna。
[0048]
在本发明中,肝癌早期患者的粪便样本为从医学上确诊的肝癌早期患者处采集。健康人群的粪便样本为医学鉴定的健康人中采集。本发明对提取粪便中细菌dna的方法没有特殊限制,采用本领域所熟知的细菌dna提取方法即可,例如dna提取试剂盒法进行。在本发明实施例中,提取dna所用试剂盒购自广州赛百纯生物科技有限公司。
[0049]
得到健康人群组的细菌dna和肝癌早期患者组的细菌dna,本发明对所述健康人群组的细菌dna和肝癌早期患者组的细菌dna分别进行16s dna扩增、文库构建和16s rrna测序,对测序数据进行生物信息学分析,得到两组样本中微生物属水平间的丰度差异。
[0050]
在本发明中,16s rrna扩增用引物为所述检测试剂,为了便于后续文库构建,所述检测试剂优选带接头,例如341f(seq id no:53)和805r(seq id no:54),其中划线部分为接头引物序列,非划线部分为16s v3v4区域序列,兼并碱基n=a、g、c或t,w=a或t,h=a、c或t,v=a、g或c。所述16s dna扩增的反应程序优选如下:95℃3min;95℃30sec;55℃30sec;72℃30sec,25个循环;72℃5min。扩增结束后优选进行纯化,本发明对所述纯化的方法没有特殊限制,采用本领域所熟知的纯化方法即可。
[0051]
本发明对所述文库构建的方法没有特殊限制,采用本领域所熟知的文库构建的方法即可。所述文库构建时优选对纯化后的pcr产物进行扩增。所述扩增用的引物优选包括fwd和rev;所述fwd的核苷酸序列如seq id no:55所示和rev的核苷酸序列如seq id no:56所示,其中[i5]和[i7]是标签序列,仅用于区分每个样本。文库扩增的反应程序优选如下:95℃3min;95℃30sec;55℃30sec;72℃30sec,8循环;72℃5min。
[0052]
在本发明中,构建的文库优选适合illuminamiseq测序技术。优选使用illuminamiseq仪器对合并的文库进行测序,测序试剂盒采用miseq reagent kit v3(illumina,inc.,san diego,ca,usa)。
[0053]
在本发明中,所述生物信息学分析的方法优选为通过dada2对测序数据进行过滤、去噪、拼接、去嵌合体形成16s rrna基因的序列变异体,参考silva数据库对所述序列变异体进行分类注释,并通过stamp中welch
‘
s t
‑
test检验分析(p=0.05)肝癌早期患者和健康人群中肠道微生物属水平间的丰度差异。得到布劳特氏菌属(blautia)、大肠埃希菌
‑
志贺氏菌属(escherichia
‑
shigella)、扭链瘤胃球菌(ruminococcus torques group)、链球菌属(streptococcus)、活泼瘤胃球菌(ruminococcus gnavus group)、霍氏真杆菌(eubacterium hallii group)、多尔氏菌属(dorea)在肝癌早期人群的粪便中相对丰度显
著升高;拟杆菌属(bacteroides)、普雷沃氏菌属9(prevotella 9)、毛螺菌属(lachnospira)、挑剔真杆菌(eubacterium eligens group)、粪杆菌属(faecalibacterium)、考拉杆菌属(phascolarctobacterium)、巨单胞菌属(megamonas)、lachnospiraceae nk4a136 group在肝癌早期人群的粪便中相对丰度显著降低。
[0054]
在本发明中,筛选有显著差异的生物标志物的方法优选为用线性判别分析和效应大小方法进行筛选。
[0055]
在本发明中,通过所述生物标志物建立特异性鉴定肝癌早期和健康人群样本的随机森林模型的方法优选为将全部肝癌早期样本和健康人群样本分别按数量比4:1分为训练集和隔离集,使用python软件sklearn.ensemble.randomforestclassifier模块对所述训练集进行随机森林模型训练。所述随机森林模型训练优选包括使用网格搜索方法调整模型训练参数,用十次交叉验证训练随机森林模型;
[0056]
所述模型训练参数设定如下:梯度n_estimators=[100,500,1000];max_depth=[1,2,3,7,9];max_features=["log2","sqrt"];
[0057]
所述十次交叉验证训练随机森林模型的方法为使用python软件sklearn.model_selection.gridsearchcv模块搜索最佳参数,共计30种参数组合,每种参数组合下将训练集分割成10份子样本,一份单独的子样本被保留作为验证模型的数据,其他9份子样本用来训练;重复10次,每份子样本验证一次,平均10次的结果作为该参数组合下最终模型结果;总计模型训练次数为300次。
[0058]
在本发明中,所述评估的方法优选包括通过绘制学习曲线评估模型的拟合情况,同时通过接受者操作特性曲线评估模型效果,所述隔离集用于最终模型准确性评估。学习曲线评估模型无过拟合和欠拟合情况。模型的曲线下面积为0.95。实验证明,用这14种生物标志物构建的预测模型具有区分肝癌和健康样本的能力。
[0059]
在本发明中,进一步评估生物标志物建立预测模型对肝癌早期患者和健康对照的诊断能力。采用28例肝癌早期患者和39例健康人群)验证训练出的随机森林模型用于肝癌早期诊断的准确性,结果表明,测试的回归结果精度为0.97,召回率为1.0,特异度为0.98。本发明的生物标记物、随机森林训练模型以及检测方法可用于肝癌早期诊断。
[0060]
下面结合实施例对本发明提供的一种用于原发性肝细胞癌早期诊断的生物标志物及其检测试剂和应用进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
[0061]
实施例1
[0062]
肝癌早期肠道生物标志物的筛选
[0063]
一、肠道细菌的dna提取
[0064]
从广西地区搜集肝癌早期患者粪便样本141例,健康人群对照粪便样本194例,用于筛选肝癌早期肠道生物标志物,具体步骤如下:
[0065]
1:上述的粪便样本分装样本并冻存于
‑
80℃;
[0066]
2:提取s1所述肝癌患者和健康人群的粪便细菌dna;
[0067]
3:对粪便细菌dna进行16s rrna扩增和文库构建,具体步骤如下:
[0068]
s1、核酸提取(surbiopure粪便核酸提取试剂盒(磁珠法),广州赛百纯生物科技有限公司)。
[0069]
s2.取0.25g粪便样本(同时设置一空白对照与mock对照)加入dry beads tube中,加入900μl s1
‑
lysis enhancer涡旋彻底、混匀。
[0070]
s3.加入100μl s2
‑
lysis enhancer溶液至样品中,65℃孵育10min。
[0071]
s4.剧烈涡旋震荡10min。
[0072]
s5.12000 rpm离心5min,转移上清600μl到新的1.5ml的离心管中。
[0073]
s6.加入400μl s3
‑
cleanup buffer,立即彻底混匀。
[0074]
s7.12000 rpm离心2min,转移全部上清液到预分装板的孔中。使用核酸自动提取仪(gene pure核酸提取仪,上海宝予德科学仪器有限公司)进行提取。
[0075]
s8.将提取的dna至新的离心管
‑
20℃保存。
[0076]
s9.使用超微量分光光度计colibri lb 915(brethold technologies)对dna纯度进行质控。
[0077]
二、文库构建和测序
[0078]
对提取的dna使用qubittm4.0(thermo fisher scientifi)测量的每1μl dna样品的浓度约为10~100ng/μl,使用transstartfastpfu fly dna polymerase试剂盒(北京全式金生物技术(trans gen biotech)有限公司)通过扩增仪miniamp plus thermal cycler(thermo fisher scientifi)扩增细菌16s rrna基因的v3~v4区,引物组为带接头的341f(5
’‑
tcgtcggcagcgtcagatgtgtataagagacagcctacgggnggcwgcag
‑3’
,seq id no:53)和805r(5
’‑
gtctcgtgggctcggagatgtgtataagagacaggactachvgggtatctaatcc
‑3’
,seq id no:54,其中划线部分为接头引物序列,非划线部分为16s v3v4区域序列,兼并碱基n=a,g,c,t,w=a,t,h=a,c,t,v=a,g,c),反应体系如表2所示。
[0079]
表2 16s rrna基因的pcr扩增反应体系
[0080]
试剂体积(ul)细菌dna(1ng/μl)1341f10μm0.5805r10μm0.5fly聚合酶0.5缓冲液(5
×
)5dntp2ddh2o15.5总计25
[0081]
16s rrna基因的pcr扩增反应程序:95℃3min;95℃30sec;55℃30sec;72℃30sec,25个循环;72℃5min。
[0082]
使用凝胶成像仪bioanalytical imaging system(azure biosystems)根据pcr产物的大小验证扩增的dna后,使用磁珠法(magnetic dna beads,北京全式金生物技术(transgen biotech)有限公司)进行纯化。
[0083]
使用适合illumina miseq的标签引物对上述pcr纯化产物进行文库扩增,引物为fwd(5
’‑
aatgatacggcgaccaccgagatctacac[i5]tcgtcggcagcgtc
‑3’
,seq id no:55)和rev(5
’‑
caagcagaagacggcatacgagat[i7]gtctcgtgggctcgg
‑3’
,seq id no:56),反应体系见表3。
[0084]
表3文库扩增的pcr扩增体系
[0085]
试剂体积(μl)dna(1ng/μl)1μlfwd引物0.5μlrev引物0.5μlfly聚合酶0.25μlbuffer(5
×
)2.5μldntp0.5μlddh2o7.25μl总计12.5μl
[0086]
文库扩增的pcr反应程序如下:95℃3min;95℃30sec;55℃30sec;72℃30sec,8个循环;72℃5min。
[0087]
pcr扩增结束后,使用磁珠法(magnetic dna beads,北京全式金生物技术(transgen biotech)有限公司)纯化pcr产物。使用生物分析仪agilent 2100(agilent technologies)和qubit
tm
4.0(thermo fisher scientifi)对纯化的产物进行定量。使用illuminamiseq仪器对合并的文库进行测序,测序试剂盒采用miseq reagent kit v3(illumina,inc.,san diego,ca,usa);随后在illumina miseq测序平台完成16s rrna测序,并对测序文件进行质控过滤和生物信息学分析,主要通过dada2对测序文件进行过滤、去噪、拼接、去嵌合体形成16s rrna基因的序列变异体(过滤的参数为
‑‑
p
‑
trunc
‑
q 2,降噪参数为
‑‑
p
‑
pooling
‑
method'independent',合并的参数为
‑‑
p
‑
trunc
‑
len
‑
f 260,
‑‑
p
‑
trunc
‑
len
‑
r 220,overlap=12,去嵌合体的参数为
‑‑
p
‑
chimera
‑
method'consensus',
‑‑
p
‑
min
‑
fold
‑
parent
‑
over
‑
abundance 1),参考silva数据库对序列变异体进行分类注释(参见quast c,pruesse e,yilmaz p,gerken j,schweer t,yarza p,peplies j,fo.the silvaribosomal rna gene database project:improved data processing andweb
‑
basedtools.nucleicacids res.2013jan;41(database issue):d590
‑
6.doi:10.1093/nar/gks1219.epub 2012 nov 28.pmid:23193283;pmcid:pmc3531112.),并通过stamp(参见parks dh,tyson gw,hugenholtz p,beiko rg.stamp:statistical analysis oftaxonomic and functionalprofiles.bioinformatics.2014 nov 1;30(21):3123
‑
4.doi:10.1093/bioinformatics/btu494.epub 2014 jul 23.pmid:25061070;pmcid:pmc4609014.)中welch
‘
s t
‑
test检验(p=0.05)分析肝癌早期患者和健康对照的肠道微生物属水平间的丰度差异。
[0088]
三、使用线性判别分析和效应大小方法(lefse,具体参见segatan,izardj,waldron l,gevers d,miropolsky l,garrettws,huttenhower c.metagenomic biomarker discovery and explanation.genome biol.2011jun 24;12(6):r60.doi:10.1186/gb
‑
2011
‑
12
‑6‑
r60.pmid:21702898;pmcid:pmc3218848.)找到组间有显著差异的生物标志物。
[0089]
结果:根据上述方法,找到了14个菌属在肝癌患者和健康人群间变化显著的生物标志物,包括如下菌属:布劳特氏菌属(blautia)、大肠埃希菌
‑
志贺氏菌属(escherichia
‑
shigella)、扭链瘤胃球菌(ruminococcus torques group)、链球菌属(streptococcus)、活泼瘤胃球菌(ruminococcus gnavus group)、霍氏真杆菌(eubacterium hallii group)、多
尔氏菌属(dorea)、拟杆菌属(bacteroides)、普雷沃氏菌属9(prevotella 9)、毛螺菌属(lachnospira)、挑剔真杆菌(eubacterium eligens group)、粪杆菌属(faecalibacterium)、考拉杆菌属(phascolarctobacterium)、巨单胞菌属(megamonas)、lachnospiraceae nk4a136 group,如图1所示。其中,布劳特氏菌属(blautia)、大肠埃希菌
‑
志贺氏菌属(escherichia
‑
shigella)、扭链瘤胃球菌(ruminococcus torques group)、链球菌属(streptococcus)、活泼瘤胃球菌(ruminococcus gnavus group)、霍氏真杆菌(eubacterium hallii group)、多尔氏菌属(dorea)在肝癌早期人群的粪便中相对丰度显著升高;拟杆菌属(bacteroides)、普雷沃氏菌属9(prevotella 9)、毛螺菌属(lachnospira)、挑剔真杆菌(eubacterium eligens group)、粪杆菌属(faecalibacterium)、考拉杆菌属(phascolarctobacterium)、巨单胞菌属(megamonas)、lachnospiraceae nk4a136 group在肝癌早期人群的粪便中相对丰度显著降低,如图2所示。
[0090]
实施例2
[0091]
利用生物标志物建立预测模型对肝癌早期患者和健康对照的区分能力
[0092]
期望通过以上找到的15种肠道生物标志物建立一个能够特异性鉴定肝癌早期和健康人群样本的随机森林模型。采集了113例肝癌早期和155例健康对照的粪便,采取与实施例1相同的处理方法,使用网格搜索方法调整模型训练参数并进行十次交叉验证对这268例粪便的15种肠道生物标志物训练随机森林模型,具体为设置随机森林模型参数梯度n_estimators=[100,500,1000];max_depth=[1,2,3,7,9];max_features=["log2","sqrt"]。使用python软件sklearn.model_selection.gridsearchcv模块搜索最佳参数,共计3*5*2=30种参数组合,每种参数组合下将训练集分割成10份子样本,一份单独的子样本被保留作为验证模型的数据,其他9份子样本用来训练。重复10次,每份子样本验证一次,平均10次的结果作为该参数组合下最终模型结果。总计模型训练次数为300次(30*10)。绘制学习曲线评估模型的拟合情况,通过接受者操作特性(receiver operating characteristic,roc)曲线评估模型效果。
[0093]
结果:一方面,通过学习曲线来评估模型的过拟合和欠拟合情况。训练集准确率与验证集准确率随着样本量增加而收敛,但收敛后的准确率远小于期望的准确率(desired accuracy=1.0),即训练集和验证集的准确率具有较高偏差,说明模型欠拟合;模型在训练集有很高的的准确率而在验证集中有较低准确率,训练集和测试集的准确率有较大差距时,为高方差,说明模型过拟合。如图3所示,模型训练集和测试集都有较高的准确率,且随着样本量增加验证集准确率趋于水平,说明该模型结果较好。
[0094]
另一方面,通过roc曲线描述在各种不同阈值下真正率(true positive rate,tpr)相对于假正率(false positive rate,fpr)的取值变化情况来评估模型的效果。在机器学习中,tpr被称为灵敏度(sensitivuty)或者召回率(eecall),而fpr被称为fall
‑
out或虚警率(probability of false alarm),具体计算说明见表4。roc曲线的横坐标是假阳性比值(假正率),纵坐标是真阳性比值(真正率),假正率反应了模型虚报的响应程度,真正率反应了模型预测响应的覆盖程度。希望假正率越低,真正率越高,模型就越好。反应到roc图形上,也就是曲线越陡峭,越朝着左上方突出,模型效果越好。我们通过计算曲线下面积(area under curve,auc)值评估模型的预测效果,在roc曲线图上,如果我们连接对角线,
它的面积正好是0.5,对角线的实际含义是:随机判断响应与不响应,正负样本覆盖率应该都是50%,表示随机效果。roc曲线越陡越好,所以理想auc值=1(参见tom fawcett.2006.an introduction to roc analysis.pattern recogn.lett.27,8(june 2006),861
–
874.doi:https://doi.org/10.1016/j.patrec.2005.10.010)。如图4所示,模型的roc曲线auc值为0.95。证明用这15种标志物构建的模型具有可以区分肝癌早期和健康样本的能力。
[0095]
表4模型的混淆矩阵说明
[0096] 测试值(早期癌症)测试值(健康)真实值(早期癌症)tpfn真实值(健康)fptn
[0097]
注:tp:true postive,真阳性:被模型预测成癌症的癌症样本;
[0098]
fp:false positive,假阳性:被模型预测成癌症的健康样本;
[0099]
fn:false negative,假阴性:被模型预测成健康的癌症样本;
[0100]
tn:true negative,真阴性:被模型预测成健康的健康样本;
[0101]
精度(precision)=查全率=tp/(tp+fp),是指在所有被预测为癌症的样本中实际为癌症样本的概率;
[0102]
特异度=tn/(tn+fp),指在实际为健康样本预测成健康样本的概率;
[0103]
真正率(tpr)=灵敏度=召回率=查全率=tp/(tp+fn),是指在实际为癌症的样本中被预测为癌症样本的概率;
[0104]
假正率(fpr)=1
‑
特异度=fp/(tn+fp),即实际为健康样本预测成癌症样本的概率。
[0105]
实施例3
[0106]
评估生物标志物建立预测模型对肝癌早期患者和健康对照的诊断能力
[0107]
使用模型训练中分离出的隔离集数据(包括28例肝癌早期患者和39例健康人群)验证训练出的随机森林模型用于肝癌早期诊断的准确性。采取与实施例1相同的处理方式,欲验证筛选出的生物标记物和训练出的随机森林模型可用于肝癌早期诊断。
[0108]
结果:混淆矩阵的结果表5,测试的回归结果精度为0.97,召回率为1.0,特异度为0.98。由此可见,本发明的生物标记物、随机森林训练模型以及检测方法可用于肝癌早期诊断。
[0109]
表5 28例肝癌早期患者和39例健康人群对模型的测试结果
[0110] 测试值(早期癌症)测试值(健康)真实值(早期癌症,n=28)tp=28fn=0真实值(健康,n=39)fp=1tn=38
[0111]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1