一种高纯度ssDNA制备方法与流程
本发明属于分子生物学技术领域,涉及一种ssdna制备方法,具体为一种高纯度ssdna制备方法。
背景技术:
基因编辑是一种新兴的比较精确的能对生物体基因组特定目标基因进行修饰的一种基因工程技术。基因编辑技术的最新发展已经改变了我们操作基因组的能力。可编辑位点特异性核酸酶,特别是crispr/cas系统,在目标基因组位置引入双链断裂,然后可以通过选择内源性dna进行工程机制修复。
crispr/cas9基因编辑工具已经被成功应用于获得基因敲除突变(knockout),但众多事实证明在基因敲入上的应用有很大的难度。基因敲入所面临的主要难题是修复模板的制备和如何与cas9-sgrna核糖核蛋白复合物共导入至细胞。ssdna常作为“donortemplate”可实现定点修复和长片段的敲入。研究表明ssdna作为同源臂具有更高的定点修复和大片段敲入的效率,更易筛选获得定点修复和大片段的敲入的突变体。相比于双链dna模板具有显著降低基因组随机整合的概率,提高基因编辑效率和更低细胞毒性的优点。但是其制备难度和制备费用都有很大的挑战性。
迄今为止,体外制备单链ssdna的方法有很多种:例如化学合成;不对称pcr;利用链霉亲和素颗粒分离亲和素标记单链;lambda核酸外切酶消化磷酸化单链;滚环pcr扩增;限制性内切酶剪切质粒后变性等。但这些方法还是会出现混入大量dsdna的情况。
技术实现要素:
本发明的目的就在于为了解决迄今为止,体外制备单链ssdna的方法有很多种:例如化学合成;不对称pcr;利用链霉亲和素颗粒分离亲和素标记单链;lambda核酸外切酶消化磷酸化单链;滚环pcr扩增;限制性内切酶剪切质粒后变性等。但这些方法还是会出现混入大量dsdna的情况,而本发明提供了一种去除大量dsdna的方法,先是利用lambda核酸外切酶消化磷酸化单链的方法得到ssdna,然后在通过双链特异性酶去除残留双链dna,得到了高纯度无dsdna残留的ssdna,克服了残留大量dsdna的问题。
本发明的目的可以通过以下技术方案实现:
一种高纯度ssdna制备方法,包括以下步骤:
第一步:合成所需目的基因到puc57载体中;
第二步:磷酸化双链dna的合成;
第三步:lambda核酸外切酶消化磷酸化双链dna,得到ssdna;
第四步:通过双链特异性酶去除残留双链dna,得到高纯度的ssdna。
优选的,合成所需目的基因到puc57载体中具体包括以下步骤:
第一步:根据需要ssdna序列,通过重叠pcr的原理扩增得到所需dna序列,得到pcr产物;
第二步:将puc57载体质粒通过ecorv酶使载体线性化,将pcr产物与线性化载体连接,得到重组产物;
第三步:将重组产物取9-11ul转移到t1感受态细胞中,置于冰上8-12min,然后40-44℃热激85-95s,再置于冰上1-5min,加入550-650ullb培养基,35-39℃,210-230rpm条件下培养38-42min,取90-110ul涂平板,最后37℃过夜培养;
第四步:第二天,利用菌落pcr方法筛选阳性克隆,并利用sanger法测序验证目的序列完全正确,得到正确克隆。
优选的,磷酸化双链dna的合成具体包括以下步骤:
s1:合成尾引物为磷酸化引物,首引物为普通引物,pcr扩增目的基因;
pcr体系如下:工作液90-94ul、引物f1-3ul、磷酸化引物r1-3ul、模板0.4-0.6ul、a酶1-3ul;pcr程序如下:先于98℃保持10s进行变性,然后将温度降至58℃保持25s进行退火,再在72℃保持30s进行延伸,合成磷酸化双链dna,完成一组循环,重复循环30次,在温度72℃保持2min,最后在4℃保存;
s2、琼脂糖凝胶电泳检测扩增条带是否单一清晰,与目的基因大小一致,利用pcra回收试剂盒回收pcr产物,否则弃用;回收产物用去核酸酶水洗脱,浓度应在100-200ng/ul。
优选的,利用lambda核酸外切酶消化磷酸化双链dna,得到ssdna;反应体系如下:10×buffer1-3ul、dsdna1-2ug、lambda酶1-1.5ul、nfh2o14.5-15ul;反应条件如下:37℃5min,80℃5min,4℃1min。
优选的,通过双链特异性酶去除残留双链dna的反应体系为ssdna产物16-18ul、10×dsdnasebuffer1-3ul、dsdnase0.8-1.2ul;反应条件如下:37℃5min,80℃5min;酶切后的ssdna通过异丙醇沉淀的方法得到无双链残留、高纯度的ssdna;再利用s1酶验证dsdna是否消化完全。
与现有技术相比,本发明的有益效果是:本发明提供了一种去除大量dsdna的方法,先是利用lambda核酸外切酶消化磷酸化单链的方法得到ssdna,然后在通过双链特异性酶去除残留双链dna,得到了高纯度无dsdna残留的ssdna,克服了残留大量dsdna的问题。
附图说明
为了便于本领域技术人员理解,下面结合附图对本发明作进一步的说明。
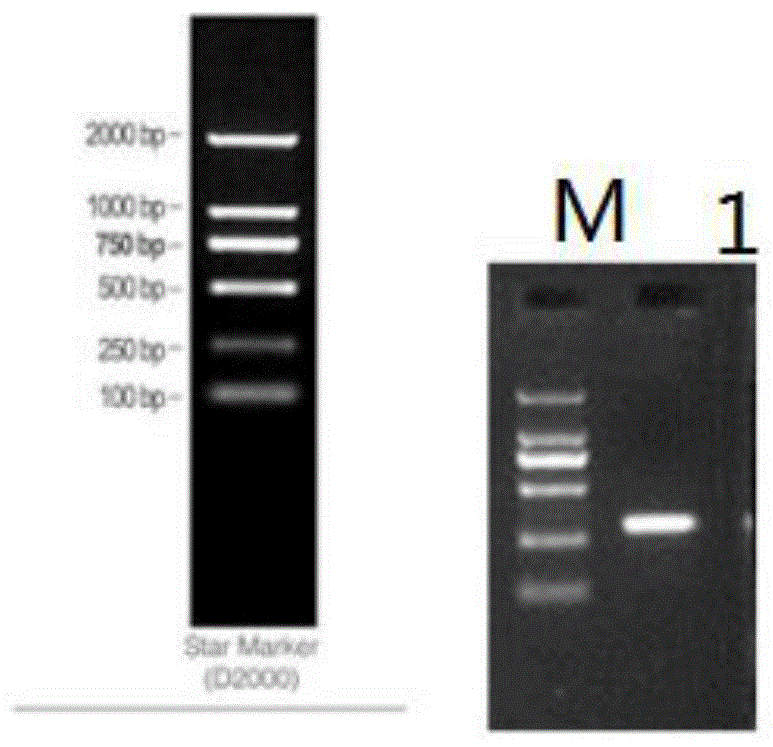
图1为本发明的实施例中磷酸化dsdna凝胶电泳检测图,泳道m为marker,泳道1为300bpdsdna。
图2为本发明的实施例中未利用双链特异性酶处理的ssdna凝胶电泳检测图,泳道m为marker,泳道1为300bpssdna,泳道2为用s1酶消化后的产物。
图3为本发明的实施例中利用双链特异性酶处理后的ssdna凝胶电泳检测图,泳道m为marker,泳道1为300bpssdna,泳道2为用s1酶消化后的产物。
具体实施方式
下面将结合实施例对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
实施例1
请参阅图1-3所示,一种高纯度ssdna制备方法,包括以下步骤:
第一步:合成所需目的基因到puc57载体中;
第二步:磷酸化双链dna的合成;
第三步:lambda核酸外切酶消化磷酸化双链dna,得到ssdna;
第四步:通过双链特异性酶去除残留双链dna,得到高纯度的ssdna。
优选的,合成所需目的基因到puc57载体中具体包括以下步骤:
第一步:根据需要ssdna序列,通过重叠pcr的原理扩增得到所需dna序列,得到pcr产物;
第二步:将puc57载体质粒通过ecorv酶使载体线性化,将pcr产物与线性化载体连接,得到重组产物;
第三步:将重组产物取9ul转移到t1感受态细胞中,置于冰上8min,然后40℃热激85s,再置于冰上1min,加入550ullb培养基,35℃,210rpm条件下培养38min,取90ul涂平板,最后37℃过夜培养;
第四步:第二天,利用菌落pcr方法筛选阳性克隆,并利用sanger法测序验证目的序列完全正确,得到正确克隆。
磷酸化双链dna的合成具体包括以下步骤:
s1:合成尾引物为磷酸化引物,首引物为普通引物,pcr扩增目的基因;
pcr体系如下:工作液90ul、引物f1ul、磷酸化引物r1ul、模板0.4ul、a酶1ul;pcr程序如下:先于98℃保持10s进行变性,然后将温度降至58℃保持25s进行退火,再在72℃保持30s进行延伸,合成磷酸化双链dna,完成一组循环,重复循环30次,在温度72℃保持2min,最后在4℃保存;
s2、琼脂糖凝胶电泳检测扩增条带是否单一清晰,与目的基因大小一致,利用pcra回收试剂盒回收pcr产物,否则弃用;回收产物用去核酸酶水洗脱,浓度应在100ng/ul。
利用lambda核酸外切酶消化磷酸化双链dna,得到ssdna;反应体系如下:10×buffer1ul、dsdna1ug、lambda酶1ul、nfh2o14.5ul;反应条件如下:37℃5min,80℃5min,4℃1min。
通过双链特异性酶去除残留双链dna的反应体系为ssdna产物16ul、10×dsdnasebuffer1ul、dsdnase0.8ul;反应条件如下:37℃5min,80℃5min;酶切后的ssdna通过异丙醇沉淀的方法得到无双链残留、高纯度的ssdna;再利用s1酶验证dsdna是否消化完全。
实施例2
一种高纯度ssdna制备方法,包括以下步骤:
第一步:合成所需目的基因到puc57载体中;
第二步:磷酸化双链dna的合成;
第三步:lambda核酸外切酶消化磷酸化双链dna,得到ssdna;
第四步:通过双链特异性酶去除残留双链dna,得到高纯度的ssdna。
优选的,合成所需目的基因到puc57载体中具体包括以下步骤:
第一步:根据需要ssdna序列,通过重叠pcr的原理扩增得到所需dna序列,得到pcr产物;
第二步:将puc57载体质粒通过ecorv酶使载体线性化,将pcr产物与线性化载体连接,得到重组产物;
第三步:将重组产物取10ul转移到t1感受态细胞中,置于冰上10min,然后42℃热激90s,再置于冰上3min,加入600ullb培养基,37℃,220rpm条件下培养40min,取100ul涂平板,最后37℃过夜培养;
第四步:第二天,利用菌落pcr方法筛选阳性克隆,并利用sanger法测序验证目的序列完全正确,得到正确克隆。
磷酸化双链dna的合成具体包括以下步骤:
s1:合成尾引物为磷酸化引物,首引物为普通引物,pcr扩增目的基因;
pcr体系如下:工作液92ul、引物f2ul、磷酸化引物r2ul、模板0.5ul、a酶2ul;pcr程序如下:先于98℃保持10s进行变性,然后将温度降至58℃保持25s进行退火,再在72℃保持30s进行延伸,合成磷酸化双链dna,完成一组循环,重复循环30次,在温度72℃保持2min,最后在4℃保存;
s2、琼脂糖凝胶电泳检测扩增条带是否单一清晰,与目的基因大小一致,利用pcra回收试剂盒回收pcr产物,否则弃用;回收产物用去核酸酶水洗脱,浓度应在150ng/ul。
利用lambda核酸外切酶消化磷酸化双链dna,得到ssdna;反应体系如下:10×buffer2ul、dsdna2ug、lambda酶1.5ul、nfh2o15ul;反应条件如下:37℃5min,80℃5min,4℃1min。
通过双链特异性酶去除残留双链dna的反应体系为ssdna产物17ul、10×dsdnasebuffer2ul、dsdnase1.0ul;反应条件如下:37℃5min,80℃5min;酶切后的ssdna通过异丙醇沉淀的方法得到无双链残留、高纯度的ssdna;再利用s1酶验证dsdna是否消化完全。
实施例3
一种高纯度ssdna制备方法,包括以下步骤:
第一步:合成所需目的基因到puc57载体中;
第二步:磷酸化双链dna的合成;
第三步:lambda核酸外切酶消化磷酸化双链dna,得到ssdna;
第四步:通过双链特异性酶去除残留双链dna,得到高纯度的ssdna。
优选的,合成所需目的基因到puc57载体中具体包括以下步骤:
第一步:根据需要ssdna序列,通过重叠pcr的原理扩增得到所需dna序列,得到pcr产物;
第二步:将puc57载体质粒通过ecorv酶使载体线性化,将pcr产物与线性化载体连接,得到重组产物;
第三步:将重组产物取11ul转移到t1感受态细胞中,置于冰上12min,然后44℃热激95s,再置于冰上5min,加入650ullb培养基,39℃,230rpm条件下培养42min,取110ul涂平板,最后37℃过夜培养;
第四步:第二天,利用菌落pcr方法筛选阳性克隆,并利用sanger法测序验证目的序列完全正确,得到正确克隆。
磷酸化双链dna的合成具体包括以下步骤:
s1:合成尾引物为磷酸化引物,首引物为普通引物,pcr扩增目的基因;
pcr体系如下:工作液94ul、引物f3ul、磷酸化引物r3ul、模板0.6ul、a酶ul;pcr程序如下:先于98℃保持10s进行变性,然后将温度降至58℃保持25s进行退火,再在72℃保持30s进行延伸,合成磷酸化双链dna,完成一组循环,重复循环30次,在温度72℃保持2min,最后在4℃保存;
s2、琼脂糖凝胶电泳检测扩增条带是否单一清晰,与目的基因大小一致,利用pcra回收试剂盒回收pcr产物,否则弃用;回收产物用去核酸酶水洗脱,浓度应在200ng/ul。
利用lambda核酸外切酶消化磷酸化双链dna,得到ssdna;反应体系如下:10×buffer2ul、dsdna2ug、lambda酶1.5ul、nfh2o15ul;反应条件如下:37℃5min,80℃5min,4℃1min。
通过双链特异性酶去除残留双链dna的反应体系为ssdna产物18ul、10×dsdnasebuffer3ul、dsdnase1.2ul;反应条件如下:37℃5min,80℃5min;酶切后的ssdna通过异丙醇沉淀的方法得到无双链残留、高纯度的ssdna;再利用s1酶验证dsdna是否消化完全。
以上公开的本发明优选实施例只是用于帮助阐述本发明。优选实施例并没有详尽叙述所有的细节,也不限制该发明仅为的具体实施方式。显然,根据本说明书的内容,可作很多的修改和变化。本说明书选取并具体描述这些实施例,是为了更好地解释本发明的原理和实际应用,从而使所属技术领域技术人员能很好地理解和利用本发明。本发明仅受权利要求书及其全部范围和等效物的限制。
- 还没有人留言评论。精彩留言会获得点赞!