一种菲类化合物、制备方法及应用
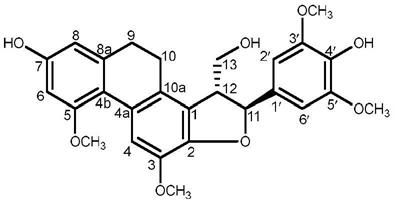
1.本发明属于一种天然提取物及其制备方法和应用,具体涉及一种菲类化合物、制备方法及应用。
背景技术:
2.许多因素均可引起炎症反应,如生物因素(细菌、病毒等)、物理因素(高温、低温、紫外线等)、化学因素(强酸、强碱、芥子气等)、异物和坏死组织等,若不及时治疗,尤其是长期慢性炎症常会导致多种疾病,亦有实验证明,目前成为非传染性流行病的糖尿病就与长期反复的慢性炎症有关。
3.目前,临床上应用的抗炎药以化药为主,主要分为甾体类和非甾体类抗炎药,其中,甾体类抗炎药有很大的副作用,应尽量少用,而非甾体抗炎药虽相对安全,也有很多仍然存在较大的副作用,如最常用的阿司匹林,就有较为严重的胃肠道反应。
技术实现要素:
4.本发明的主要目的是解决目前临床上应用的化药类抗炎药存在较大副作用的技术问题,提供一种菲类化合物、制备方法及应用。
5.为实现上述目的,本发明提供如下技术方案:
6.一种菲类化合物,其特殊之处在于,所述菲类化合物的结构式为
[0007][0008]
所述菲类化合物的分子式为c
27
h
28
o8。
[0009]
同时,本发明公开了一种上述菲类化合物的提取分离方法,其特殊之处在于,包括以下步骤:
[0010]
s1,取合叶子,通过室温冷浸或加热回流提取至少一次,将每一次的提取液合并,经过浓缩后得到总浸膏;
[0011]
s2,将经步骤s1得到的总浸膏脱脂萃取后,分离除去有机层,对水层进行有机溶剂萃取,得到萃取层;
[0012]
s3,将经步骤s2得到的萃取层上样于硅胶柱进行梯度洗脱,分别以石油醚
‑
乙酸乙酯和二氯甲烷
‑
甲醇系统作为洗脱液,进行梯度洗脱,收集并合并相同流份,共得到17个流分;
[0013]
s4,将经步骤s3得到的17个流份中的第11个流份经硅胶柱色谱,以二氯甲烷
‑
甲醇进行梯度洗脱,收集并合并相同流份,得到3个流份;
[0014]
s5,对经步骤s4得到的3个流份中的第3个流份经硅胶柱色谱,以二氯甲烷
‑
甲醇进
行等度洗脱,收集并合并相同流份,得到3个流份;
[0015]
s6,对经步骤s5得到的3个流份中的第2个流份经凝胶柱色谱进行分离,收集并合并相同流份,得到5个流份;
[0016]
s7,对经步骤s6得到的5个流份中的第3个流份经高效液相色谱进行分离,得到菲类化合物。
[0017]
进一步地,步骤s2中,所述脱脂萃取具体为通过石油醚脱脂后,再经乙酸乙酯进行至少一次萃取。
[0018]
进一步地,步骤s4中,所述以二氯甲烷
‑
甲醇进行等度洗脱,具体是以二氯甲烷
‑
甲醇按照20:1,10:1,5:1,v/v进行梯度洗脱;
[0019]
步骤s5中,所述以二氯甲烷
‑
甲醇进行等度洗脱,具体是以二氯甲烷
‑
甲醇按照25:1,v/v进行等度洗脱;
[0020]
步骤s6中,所述经凝胶柱色谱进行分离,具体是采用甲醇作为洗脱剂;
[0021]
步骤s7中,所述经高效液相色谱进行分离,具体是采用甲醇
‑
水作为流动相,按照55:45,v/v进行分离。
[0022]
进一步地,所述步骤s1具体为,
[0023]
取合叶子,采用甲醇或体积分数为85
‑
95%的乙醇作为提取剂,料液比为1kg:2
‑
8l,通过加热回流提取1
‑
3次,每次1
‑
2h,将每一次的提取液合并,经过浓缩后得到总浸膏;
[0024]
或者,取合叶子,室温冷浸提取2
‑
4次,每次4
‑
24h,将每一次的提取液合并,经过浓缩后得到总浸膏。
[0025]
进一步地,步骤s2中,所述将经步骤s1得到的总浸膏脱脂萃取,具体为将步骤s1得到的总浸膏按照总浸膏和水的体积比为1:1
‑
1:3混悬于水中,再脱脂萃取。
[0026]
进一步地,步骤s2中,所述将经步骤s1得到的总浸膏脱脂萃取,具体为对经步骤s1得到的总浸膏先用石油醚脱脂1
‑
3次,再采用超声波辅助有机溶剂的方式萃取1
‑
5次,其中,萃取时采用的有机溶剂为乙酸乙酯,总浸膏和乙酸乙酯的体积比为1:1
‑
1:3。
[0027]
另外,本发明还公开了上述菲类化合物在制备抗炎药物中的应用。
[0028]
与现有技术相比,本发明的有益效果是:
[0029]
1.本发明的菲类化合物,是首次从鄂伦春族特色药材合叶子中分离得到的一种新的菲类化合物,含有该化合物的植物合叶子分布广泛,且可人工栽培,该化合物尤其对raw264.7细胞中no的产生较好的抑制活性,有望开发为新的抗炎药物。
[0030]
2.本发明的制备方法提取分离方法简单易行,得到的化合物纯度高,制备成本低廉。
[0031]
3.本发明制备方法中采用的有机试剂来源广泛,分离提取方法简便易操作。
[0032]
4.本发明的菲类化合物经试验证明,具有抗炎作用,经试验验证对raw264.7细胞中no的产生具有良好的抑制作用,有望开发成新的抗炎药物。
附图说明
[0033]
图1为本发明实施例一制备的菲类化合物的1h
‑
nmr图谱;
[0034]
图2为本发明实施例一制备的菲类化合物的
13
c
‑
nmr图谱;
[0035]
图3为本发明实施例一制备的菲类化合物的dept135
°
图谱;
[0036]
图4为本发明实施例一制备的菲类化合物的1h
‑1h cosy图谱;
[0037]
图5为本发明实施例一制备的菲类化合物的hsqc图谱;
[0038]
图6为本发明实施例一制备的菲类化合物的hmbc图谱;
[0039]
图7为本发明实施例一制备的菲类化合物的roesy图谱;
[0040]
图8为本发明实施例一制备的菲类化合物的hr
‑
esi ms图谱。
具体实施方式
[0041]
下面将结合本发明的实施例和附图,对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例并非对本发明的限制。
[0042]
民族药是传统中药的一部分,是各少数民族人民在漫长的同疾病斗争过程中总结的宝贵经验,应该得到继承和发扬,尤其是无本民族语言记载的人数更少的少数民族药物,更应该及时得以研究和保护。合叶子(filipendula palmate pall.)为鄂伦春族人民常用的特色药材,也叫蚊子草,鄂语名称为捏母大出哈,药用部位为其干燥全草,多年生草本,高度约1米,茎有棱,近无毛,生于林缘草甸、沼泽草甸、河岸杂木灌木丛、针阔混交林下。主要用于治疗风湿、痛风、癫痫、止血、止痢、冻伤、烧伤、妇科止血等症。同时,它也常被用来作为野菜食用,以及家畜的饲料使用,是一种药食两用的植物。目前,国内外对合叶子的化学成分研究还十分有限,对其药理活性的研究报道也较少,在对其化学成分研究和药理活性筛选过程中发现了合叶子中的一个新化合物,其具有显著的抗炎活性,有望应用在抗炎药物制备中。
[0043]
本发明公开了一种从合叶子中提取菲类化合物的方法,包括以下步骤:
[0044]
1)取一定质量(kg)合叶子的干燥地上部分,用甲醇或体积分数为85
‑
95%的乙醇,在各自沸点附近加热回流提取1
‑
3次,每次1
‑
2小时,合并提取液减压回收除去溶剂,得到总浸膏,其中,合叶子的干燥地上部分与甲醇或乙醇的料液比为1kg:2
‑
8l;
[0045]
提取也可以采用室温冷浸提取,取合叶子,室温冷浸提取2
‑
4次,每次4
‑
24h,将每一次的提取液合并,经过浓缩后得到总浸膏。
[0046]
2)将总浸膏混悬于水中,总浸膏和水的体积比为1:1
‑
1:3,得到浸膏液,然后依次用等体积的有机溶剂进行多次萃取,其中,先用石油醚对总浸膏液进行等体积萃取脱脂,之后每次萃取均是分离出上一次萃取后的有机层,将剩余的水层用有机溶剂等体积进行下一次萃取,每种溶剂各萃取多次,合并萃取液,常压或减压蒸馏除去有机溶剂,分别得到各萃取层和水层。有机溶剂包括石油醚和乙酸乙酯等,且萃取次序为先用极性小的溶剂,再用极性大的有机溶剂,萃取次数及有机溶剂的选择,可根据实际操作情况进行合理调整和选择。
[0047]
3)取萃取层,通过采用柱层析纯化等分离方法,得到本发明的菲类化合物。
[0048]
柱层析包括以下五个阶段:
[0049]
第一阶段:将萃取层上样于硅胶柱,分别以石油醚
‑
乙酸乙酯和二氯甲烷
‑
甲醇系统作为洗脱液,进行梯度洗脱,收集并合并相同流份后,得到17个流份,分别编号为ea1~ea17,此部分为第一次过柱部分;
[0050]
第二阶段:将第一次过柱部分的ea11流份,其所对应的极性为二氯甲烷
‑
甲醇=5:1的流份经硅胶柱色谱进行分离,以二氯甲烷
‑
甲醇作为洗脱液,二氯甲烷
‑
甲醇(20:1,10:1,5:1,v/v)进行梯度洗脱,收集并合并相同流份,共得到3个流份,编号为ea11
‑
1~ea11
‑
3,
此部分为第二次过柱部分;
[0051]
第三阶段:将第二次过柱部分中的ea11
‑
3的流份经硅胶柱色谱,以二氯甲烷
‑
甲醇(25:1,v/v)进行等度洗脱,通过tlc检识合并相同流份后共得到3个流份,标记为ea11
‑3‑
1~ea11
‑3‑
3,此部分为第三次过柱部分;
[0052]
第四阶段:将第三次过柱部分的ea11
‑3‑
2流份经sephadex lh
‑
20凝胶柱色谱,以甲醇为洗脱剂,得到5个流份,标记为ea11
‑3‑2‑
1~ea11
‑3‑2‑
5,此部分为第四次过柱部分;
[0053]
第五阶段:将第四次过柱部分中的ea11
‑3‑2‑
3流份经hplc色谱,以甲醇
‑
水(55:45,v/v)为流动相进行洗脱,得到本发明的菲类化合物。
[0054]
上述方法制备得到的菲类化合物,其结构式如下:
[0055][0056]
本发明在对合叶子进行化学成分和药理活性的研究中发现,从中分离得到的菲类化合物对raw264.7细胞中产生的no具有显著的抑制作用,因此有望开发成新的抗炎药物或研发抗炎药物的先导化合物。
[0057]
实施例一
[0058]
(1)菲类化合物的提取和分离
[0059]
1)取干燥的合叶子地上部分15kg,用体积量为其质量3倍的95%乙醇加热回流提取3次,每次2小时,合并提取液减压回收溶剂,得总浸膏;
[0060]
2)将总浸膏混悬于3倍量的水中,先用石油醚萃取3次进行脱脂,再用乙酸乙酯等体积萃取3次,萃取层减压除去有机溶剂后得到乙酸乙酯萃取层。
[0061]
3)取乙酸乙酯萃取层155g,首先通过采用硅胶柱色谱,分别以石油醚
‑
乙酸乙酯(50:1,20:1,10:1,5:1,2:1)和二氯甲烷
‑
甲醇(20:1,10:1,5:1,3:1,2:1,1:1,1:0)进行梯度洗脱,以薄层色谱检识,合并相同流分,共得到17个流分,编号ea
‑
1~ea
‑
17。
[0062]
4)将其中的第11个流份ea
‑
11再一次经硅胶柱色谱分离,以二氯甲烷
‑
甲醇(20:1,10:1,5:1,v/v)进行梯度洗脱,每300ml收集一个流份,得到3个流份(ea
‑
11
‑
1~ea
‑
11
‑
3)。
[0063]
5)将其中第3个流份ea
‑
11
‑
3经硅胶柱色谱,以二氯甲烷
‑
甲醇(25:1,v/v)进行等度洗脱,得到3个流份(ea
‑
11
‑3‑
1~ea
‑
11
‑3‑
3)。
[0064]
6)将其中第2个流份ea
‑
11
‑3‑
2经sephadex lh
‑
20凝胶柱色谱,以甲醇为洗脱剂,得到5个流份(ea
‑
11
‑3‑2‑
1~ea
‑
11
‑3‑2‑
5)。
[0065]
7)将第3个流份ea
‑
11
‑3‑2‑
3经半制备高效液相色谱,以甲醇
‑
水(55:45,v/v)为流动相进行洗脱,得到本发明中的菲类化合物
[0066]
本发明通过理化常数和波谱技术(1d
‑
nmr,2d
‑
nmr,hr
‑
esi
‑
ms等)确定了该化合物的结构。
[0067]
(2)本发明中的菲类化合物的结构鉴定
[0068]
化合物:白色粉末,易溶于甲醇。如图8,根据hr
‑
esi ms谱,其准分子离子峰为m/z 503.16721[m+na]
+
(calcd.as 503.16764),从而确定其分子式为c
27
h
28
o8,不饱和度为14。
在1h
‑
nmr(600mhz,(cd3)2co)和
13
c
‑
nmr(150mhz,(cd3)2co)中,质子信号δ
h 2.65(m,2h),2.55(m,2h),7.85(s,1h),6.47(d,j=2.0hz,1h),6.37(d,j=2.0hz,1h),6.71(s,2h)和16个不饱和碳信号δ
c 99.1,103.8,108.2,115.1,116.4,125.5,127.3,128.2,134.4,136.3,141.3,142.7,147.4,148.7,157.3,158.5以及dept 135
°
显示的2个亚甲基信号δ
c 26.0,31.3,表明该化合物中含有一个五取代的二氢菲结构和一个四取代的苯环结构。根据质子信号δ
h 3.88(s,3h),3.87(s,3h),3.78(s,6h),8.30(s,1h),7.17(s,1h),说明该化合物中含有三个甲氧基和两个羟基。如图6所示,在hmbc谱中,h
‑
8与c
‑
10,c
‑
7,c
‑
5相关,h
‑
6与c
‑
4b相关,h
‑
4与c
‑
4b,c
‑
2,c
‑
10相关。如图7,在roesy谱中,h
‑
8与h
‑
9以及7
‑
oh上的质子相关,h
‑
6与5
‑
och3上的质子相关,h
‑
4与3
‑
och3上的质子相关,由此确定了五取代二氢菲结构上各个取代基的位置。通过质子信号δ
h 3.62(m,1h),5.74(d,j=2.8hz,1h),3.65(m,1h),3.88(m,1h)以及dept 135
°
显示的2个次甲基信号和1个亚甲基信号,推测这两个结构之间通过一个二氢呋喃环连接。在hmbc谱中,h
‑
12与c
‑
2,c
‑
10a相关,h
‑
11与c
‑
1,c
‑
2,c
‑6′
相关。在roesy谱中h
‑
13与h
‑
10相关,h
‑
11与h
‑2′
相关,因此确定c
‑
12与c
‑
1相连,c
‑
11与c
‑1′
相连,这说明两个结构之间确实通过一个二氢呋喃环相连接。此外,在hmbc谱中,h
‑2′
与c
‑
11、c
‑4′
、c
‑6′
相关,4
′‑
oh上的活泼质子与c
‑3′
、c
‑5′
相关;在roesy谱中,h
‑2′
与3
′‑
och3上的质子相关,3
′‑
och3上的质子与4
′‑
oh上的质子相关,由此确定了四取代苯环上各个取代基的位置。综合以上数据,该化合物的平面结构被确定。
[0069]
在roesy谱中,h
‑
11和h
‑
13相关,h
‑
12和h
‑2′
相关,且h
‑
11和h
‑
12的偶合常数为2.8hz,因此确定其相对构型为反式。此外,测得该化合物比旋光度为
‑
19,与文献中化合物bletillatin a(1a)的比旋光度
‑
26接近,确定c
‑
11位和c
‑
12位的绝对构型分别为s和r,由此该化合物的结构被确定,并命名为filipendutin a。是一个未见文献报道的新化合物,其结构式如下:
[0070][0071]
其核磁数据见表1,相关图谱参见图1
‑
图7;
[0072]
表1本发明中的菲类化合物的1h nmr和
13
c nmr数据
[0073][0074]
(3)抗炎实验
[0075]
实验方法:
[0076]
将raw264.7细胞接种于96孔板中,培养24h后加入脂多糖(lps)。接着不同组分别加入不同浓度的样品(本发明化合物及阳性药物吲哚美辛),空白组加入等体积溶剂。取培养不同时间后的细胞培养液50μl与griess试剂i等体积混合加入96孔板中,再加入griess试剂ii。酶标仪测定各组样品在546nm波长下的od值,利用下列公式计算no产生抑制率,并用spss软件计算被测试样品的半数抑制浓度(ic
50
值,μm)。
[0077]
no抑制率(%)=[1
–
(样品组/空白组)]
×
100%。
[0078]
实验结果:
[0079]
表2本发明中的化合物对lps刺激的raw264.7细胞产生no的抑制作用
[0080][0081]
结果分析:表2的数据表明,本发明中的菲类化合物具有很好的体外抗炎作用,对lps刺激的raw264.7细胞产生一氧化氮具有较好的抑制作用,其半数抑制浓度ic
50
为18.9
±
1.5μm,优于阳性对照药物吲哚美辛的29.4
±
2.2μm。说明本发明中的菲类化合物具有较好的抗炎作用,有望应用在抗炎药物或其先导化合物的制备中。
[0082]
实施例二
[0083]
1)取干燥的合叶子地上部分10kg,用体积量为其质量5倍的甲醇加热回流提取2次,每次2小时,合并提取液减压回收溶剂,得总浸膏;
[0084]
2)将总浸膏混悬于2倍量的水中,先用石油醚萃取3次,再用乙酸乙酯等体积萃取5次,萃取层减压除去有机溶剂后分别得到石油醚层和乙酸乙酯层。
[0085]
3)取乙酸乙酯萃取层100g,首先通过采用硅胶柱色谱,分别以石油醚
‑
乙酸乙酯(50:1,20:1,10:1,5:1,2:1)和二氯甲烷
‑
甲醇(20:1,10:1,5:1,3:1,2:1,1:1,1:0)进行梯
度洗脱,以薄层色谱检识,合并相同流分,共得到17个流分,编号ea
‑
1~ea
‑
17。
[0086]
4)其中第11个流份ea
‑
11再一次经硅胶柱色谱分离,以二氯甲烷
‑
甲醇(20:1,10:1,5:1,v/v)进行梯度洗脱,每300ml收集一个流份,得到3个流份(ea
‑
11
‑
1~ea
‑
11
‑
3)。
[0087]
5)其中第3个流份ea
‑
11
‑
3经硅胶柱色谱,以二氯甲烷
‑
甲醇(25:1,v/v)进行等度洗脱,得到3个流份(ea
‑
11
‑3‑
1~ea
‑
11
‑3‑
3)。
[0088]
6)其中第2个流份ea
‑
11
‑3‑
2经sephadex lh
‑
20凝胶柱色谱,以甲醇为洗脱剂,得到5个流份(ea
‑
11
‑3‑2‑
1~ea
‑
11
‑3‑2‑
5)。
[0089]
7)将第3个流份ea
‑
11
‑3‑2‑
3经半制备高效液相色谱,以甲醇
‑
水(55:45,v/v)为流动相进行洗脱,得到本发明中的菲类化合物
[0090]
实施例三
[0091]
1)取干燥的合叶子地上部分10kg,用体积量为其质量8倍的甲醇加热回流提取1次,提取2小时,对提取液减压回收溶剂,得总浸膏;
[0092]
2)将总浸膏混悬于1倍量的水中,先用石油醚萃取1次,再用乙酸乙酯等体积萃取2次,萃取层减压除去有机溶剂后分别得到石油醚层和乙酸乙酯层。
[0093]
3)取乙酸乙酯萃取层100g,首先通过采用硅胶柱色谱,分别以石油醚
‑
乙酸乙酯(50:1,20:1,10:1,5:1,2:1)和二氯甲烷
‑
甲醇(20:1,10:1,5:1,3:1,2:1,1:1,1:0)进行梯度洗脱,以薄层色谱检识,合并相同流分,共得到17个流分,编号ea
‑
1~ea
‑
17。
[0094]
4)其中第11个流份ea
‑
11再一次经硅胶柱色谱分离,以二氯甲烷
‑
甲醇(20:1,10:1,5:1,v/v)进行梯度洗脱,每300ml收集一个流份,得到3个流份(ea
‑
11
‑
1~ea
‑
11
‑
3)。
[0095]
5)其中第3个流份ea
‑
11
‑
3经硅胶柱色谱,以二氯甲烷
‑
甲醇(25:1,v/v)进行等度洗脱,得到3个流份(ea
‑
11
‑3‑
1~ea
‑
11
‑3‑
3)。
[0096]
6)其中第2个流份ea
‑
11
‑3‑
2经sephadex lh
‑
20凝胶柱色谱,以甲醇为洗脱剂,得到5个流份(ea
‑
11
‑3‑2‑
1~ea
‑
11
‑3‑2‑
5)。
[0097]
7)将第3个流份ea
‑
11
‑3‑2‑
3经半制备高效液相色谱,以甲醇
‑
水(55:45,v/v)为流动相进行洗脱,得到本发明中的菲类化合物
[0098]
实施例四
[0099]
1)取干燥的合叶子地上部分12kg,用体积量为其质量2倍的85%乙醇加热回流提取2次,每次提取2小时,对提取液减压回收溶剂,得总浸膏;
[0100]
2)将总浸膏混悬于2倍量的水中,先用石油醚萃取3次,再用乙酸乙酯等体积萃取5次,萃取层减压除去有机溶剂后分别得到石油醚层和乙酸乙酯层。
[0101]
3)取乙酸乙酯萃取层100g,首先通过采用硅胶柱色谱,分别以石油醚
‑
乙酸乙酯(50:1,20:1,10:1,5:1,2:1)和二氯甲烷
‑
甲醇(20:1,10:1,5:1,3:1,2:1,1:1,1:0)进行梯度洗脱,以薄层色谱检识,合并相同流分,共得到17个流分,编号ea
‑
1~ea
‑
17。
[0102]
4)其中第11个流份ea
‑
11再一次经硅胶柱色谱分离,以二氯甲烷
‑
甲醇(20:1,10:1,5:1,v/v)进行梯度洗脱,每300ml收集一个流份,得到3个流份(ea
‑
11
‑
1~ea
‑
11
‑
3)。
[0103]
5)其中第3个流份ea
‑
11
‑
3经硅胶柱色谱,以二氯甲烷
‑
甲醇(25:1,v/v)进行等度洗脱,得到3个流份(ea
‑
11
‑3‑
1~ea
‑
11
‑3‑
3)。
[0104]
6)其中第2个流份ea
‑
11
‑3‑
2经sephadex lh
‑
20凝胶柱色谱,以甲醇为洗脱剂,得到5个流份(ea
‑
11
‑3‑2‑
1~ea
‑
11
‑3‑2‑
5)。
[0105]
7)将第3个流份ea
‑
11
‑3‑2‑
3经半制备高效液相色谱,以甲醇
‑
水(55:45,v/v)为流动相进行洗脱,得到本发明中的菲类化合物
[0106]
实施例五
[0107]
1)取干燥的合叶子地上部分15kg,室温冷浸提取2次,每次10h,将每一次的提取液合并,对提取液减压回收溶剂,得总浸膏;
[0108]
2)将总浸膏混悬于2倍量的水中,先用石油醚萃取3次,再用乙酸乙酯等体积萃取5次,萃取层减压除去有机溶剂后分别得到石油醚层和乙酸乙酯层。
[0109]
3)取乙酸乙酯萃取层100g,首先通过采用硅胶柱色谱,分别以石油醚
‑
乙酸乙酯(50:1,20:1,10:1,5:1,2:1)和二氯甲烷
‑
甲醇(20:1,10:1,5:1,3:1,2:1,1:1,1:0)进行梯度洗脱,以薄层色谱检识,合并相同流分,共得到17个流分,编号ea
‑
1~ea
‑
17。
[0110]
4)其中第11个流份ea
‑
11再一次经硅胶柱色谱分离,以二氯甲烷
‑
甲醇(20:1,10:1,5:1,v/v)进行梯度洗脱,每300ml收集一个流份,得到3个流份(ea
‑
11
‑
1~ea
‑
11
‑
3)。
[0111]
5)其中第3个流份ea
‑
11
‑
3经硅胶柱色谱,以二氯甲烷
‑
甲醇(25:1,v/v)进行等度洗脱,得到3个流份(ea
‑
11
‑3‑
1~ea
‑
11
‑3‑
3)。
[0112]
6)其中第2个流份ea
‑
11
‑3‑
2经sephadex lh
‑
20凝胶柱色谱,以甲醇为洗脱剂,得到5个流份(ea
‑
11
‑3‑2‑
1~ea
‑
11
‑3‑2‑
5)。
[0113]
7)将第3个流份ea
‑
11
‑3‑2‑
3经半制备高效液相色谱,以甲醇
‑
水(55:45,v/v)为流动相进行洗脱,得到本发明中的菲类化合物
[0114]
实施例六
[0115]
1)取干燥的合叶子地上部分10kg,室温冷浸提取4次,每次24h,将每一次的提取液合并,对提取液减压回收溶剂,得总浸膏;
[0116]
2)将总浸膏混悬于2倍量的水中,先用石油醚萃取3次,再用乙酸乙酯等体积萃取5次,萃取层减压除去有机溶剂后分别得到石油醚层和乙酸乙酯层。
[0117]
3)取乙酸乙酯萃取层100g,首先通过采用硅胶柱色谱,分别以石油醚
‑
乙酸乙酯(50:1,20:1,10:1,5:1,2:1)和二氯甲烷
‑
甲醇(20:1,10:1,5:1,3:1,2:1,1:1,1:0)进行梯度洗脱,以薄层色谱检识,合并相同流分,共得到17个流分,编号ea
‑
1~ea
‑
17。
[0118]
4)其中第11个流份ea
‑
11再一次经硅胶柱色谱分离,以二氯甲烷
‑
甲醇(20:1,10:1,5:1,v/v)进行梯度洗脱,每300ml收集一个流份,得到3个流份(ea
‑
11
‑
1~ea
‑
11
‑
3)。
[0119]
5)其中第3个流份ea
‑
11
‑
3经硅胶柱色谱,以二氯甲烷
‑
甲醇(25:1,v/v)进行等度洗脱,得到3个流份(ea
‑
11
‑3‑
1~ea
‑
11
‑3‑
3)。
[0120]
6)其中第2个流份ea
‑
11
‑3‑
2经sephadex lh
‑
20凝胶柱色谱,以甲醇为洗脱剂,得到5个流份(ea
‑
11
‑3‑2‑
1~ea
‑
11
‑3‑2‑
5)。
[0121]
7)将第3个流份ea
‑
11
‑3‑2‑
3经半制备高效液相色谱,以甲醇
‑
水(55:45,v/v)为流动相进行洗脱,得到本发明中的菲类化合物
[0122]
实施例七
[0123]
1)取干燥的合叶子地上部分18kg,室温冷浸提取3次,每次4h,将每一次的提取液合并,对提取液减压回收溶剂,得总浸膏;
[0124]
2)将总浸膏混悬于2倍量的水中,先用石油醚萃取3次,再用乙酸乙酯等体积萃取5次,萃取层减压除去有机溶剂后分别得到石油醚层和乙酸乙酯层。
[0125]
3)取乙酸乙酯萃取层100g,首先通过采用硅胶柱色谱,分别以石油醚
‑
乙酸乙酯(50:1,20:1,10:1,5:1,2:1)和二氯甲烷
‑
甲醇(20:1,10:1,5:1,3:1,2:1,1:1,1:0)进行梯度洗脱,以薄层色谱检识,合并相同流分,共得到17个流分,编号ea
‑
1~ea
‑
17。
[0126]
4)其中第11个流份ea
‑
11再一次经硅胶柱色谱分离,以二氯甲烷
‑
甲醇(20:1,10:1,5:1,v/v)进行梯度洗脱,每300ml收集一个流份,得到3个流份(ea
‑
11
‑
1~ea
‑
11
‑
3)。
[0127]
5)其中第3个流份ea
‑
11
‑
3经硅胶柱色谱,以二氯甲烷
‑
甲醇(25:1,v/v)进行等度洗脱,得到3个流份(ea
‑
11
‑3‑
1~ea
‑
11
‑3‑
3)。
[0128]
6)其中第2个流份ea
‑
11
‑3‑
2经sephadex lh
‑
20凝胶柱色谱,以甲醇为洗脱剂,得到5个流份(ea
‑
11
‑3‑2‑
1~ea
‑
11
‑3‑2‑
5)。
[0129]
7)将第3个流份ea
‑
11
‑3‑2‑
3经半制备高效液相色谱,以甲醇
‑
水(55:45,v/v)为流动相进行洗脱,得到本发明中的菲类化合物
[0130]
实施例八
[0131]
1)取干燥的合叶子地上部分15kg,室温冷浸提取3次,每次16h,将每一次的提取液合并,对提取液减压回收溶剂,得总浸膏;
[0132]
2)将总浸膏混悬于2倍量的水中,先用石油醚萃取3次,再用乙酸乙酯等体积萃取5次,萃取层减压除去有机溶剂后分别得到石油醚层和乙酸乙酯层。
[0133]
3)取乙酸乙酯萃取层100g,首先通过采用硅胶柱色谱,分别以石油醚
‑
乙酸乙酯(50:1,20:1,10:1,5:1,2:1)和二氯甲烷
‑
甲醇(20:1,10:1,5:1,3:1,2:1,1:1,1:0)进行梯度洗脱,以薄层色谱检识,合并相同流分,共得到17个流分,编号ea
‑
1~ea
‑
17。
[0134]
4)其中第11个流份ea
‑
11再一次经硅胶柱色谱分离,以二氯甲烷
‑
甲醇(20:1,10:1,5:1,v/v)进行梯度洗脱,每300ml收集一个流份,得到3个流份(ea
‑
11
‑
1~ea
‑
11
‑
3)。
[0135]
5)其中第3个流份ea
‑
11
‑
3经硅胶柱色谱,以二氯甲烷
‑
甲醇(25:1,v/v)进行等度洗脱,得到3个流份(ea
‑
11
‑3‑
1~ea
‑
11
‑3‑
3)。
[0136]
6)其中第2个流份ea
‑
11
‑3‑
2经sephadex lh
‑
20凝胶柱色谱,以甲醇为洗脱剂,得到5个流份(ea
‑
11
‑3‑2‑
1~ea
‑
11
‑3‑2‑
5)。
[0137]
7)将第3个流份ea
‑
11
‑3‑2‑
3经半制备高效液相色谱,以甲醇
‑
水(55:45,v/v)为流动相进行洗脱,得到本发明中的菲类化合物
[0138]
以上所述仅为本发明的实施例,并非对本发明保护范围的限制,凡是利用本发明说明书及附图内容所作的等效结构变换,或直接或间接运用在其他相关的技术领域,均包括在本发明的专利保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1