瑞德西韦中间体的制备方法与流程
1.本发明涉及一种瑞德西韦中间体的制备方法。
背景技术:
2.瑞德西韦原是美国吉利德研发用于治疗埃博拉病毒,2020年新型新冠爆发,被用来作为新冠病毒的临床用药,美国fda已经批准了瑞德西韦上市用来治疗新冠病毒。nature(nature,2016,531,381-si(2))报道了一种瑞德西韦的制备方法,这是remdesivir二代合成方法。合成路线如下所示:
[0003][0004]
上述步骤三制备c-3(脱苄物),反应结束后,低温加入甲醇和三乙胺的甲醇溶液,升温至室温,减压浓缩,正己烷打浆后静置,倒出上清液,且重复操作三次。再加入甲醇升温至45℃后加水,浓缩剩至一定体积的水,降至室温后,析晶抽滤干燥得c-3,文献没有纯度报道,收率86%,按照上述nature中报道的后处理,加入甲醇的三乙胺淬灭后,c-3有降解的风险,而且长时间浓缩过程中,颜色会变得很深,不利于工业化放大生产。另外,中国专利cn104262345b报道了另一种制备脱苄物的后处理方法,反应结束需通过制备液相分离才能纯化,得到c-3(脱苄物)收率76%,也不利于工业化放大生产。
[0005]
上述步骤四制备c(丙叉物),反应结束后,降温并加入适量的碳酸氢钠和水终止反应。搅拌后,将混合物浓缩至干,加入乙酸乙酯和水萃取,有机层干燥浓缩后得到产物c(丙叉物),该产物为油状物,不做任何处理直接进行下一步反应。如果想要得到纯品c,通过硅胶柱柱层析才能得到。
[0006]
按照上述nature中报道的反应条件,反应结束时,若有部分的c-3(脱苄物)反应不完全,用乙酸乙酯萃取浓缩后残留在油状物中,油状物若不经过纯化,直接投入下一步反应,杂质也会参与反应,不利于工业化的放大生产。
技术实现要素:
[0007]
本发明所要解决的技术问题在于克服现有的瑞德西韦中间体的制备方法种类单一,操作复杂、不利于工业化生产的缺陷,而提供了一种瑞德西韦中间体的制备方法。本发明的制备方法操作简便、避免了柱层析分离纯化的方式,适用于工业化的放大生产。
[0008]
本发明通过以下技术方案解决上述技术问题。
[0009]
本发明提供了一种如式c所示化合物的制备方法,其包括以下步骤:
[0010]
(1)有机溶剂中,在酸的存在下,将如式c-3a或式c-3所示化合物与2,2-二甲氧基丙烷进行如下所示的反应,将反应液进行浓缩,得粗品;
[0011]
(2)将步骤(1)所得的粗品用水和二氯甲烷进行萃取,浓缩有机相,即可;
[0012][0013]
在本发明某些实施方案中,步骤(1)中,所述的有机溶剂可为本领域常规的有机溶剂,优选酮类溶剂,例如丙酮。
[0014]
在本发明某些实施方案中,步骤(1)中,所述的酸可为本领域常规的酸,又可为无机酸。所述的无机酸可为硫酸,例如浓硫酸。
[0015]
在本发明某些实施方案中,步骤(1)中,所述的2,2-二甲氧基丙烷和所述的如式c-3a或式c-3所示化合物的体积质量比可为本领域常规的体积质量比,优选1.5~3ml/g,例如1.5ml/g、2.12ml/g、2.33ml/g或3ml/g。
[0016]
在本发明某些实施方案中,步骤(1)中,所述的酸和所述的如式c-3a或式c-3所示化合物的体积质量比可为本领域常规的体积质量比,优选0.2~0.5ml/g,例如0.2ml/g、0.33ml/g或0.5ml/g。
[0017]
在本发明某些实施方案中,步骤(1)中,所述的如式c-3a或式c-3所示化合物和所述的有机溶剂的质量体积比可为本领域常规的质量体积比,优选30~50g/l,例如30g/l、37.5g/l、38.5g/l或50g/l。
[0018]
在本发明某些实施方案中,步骤(1)中,所述的反应的温度可为本领域常规的温度,优选10~60℃,例如10℃、45℃或60℃。
[0019]
在本发明某些实施方案中,步骤(1)中,所述的反应的进程可通过本领域常规的手段进行监控(例如tlc、hplc或lcms),以所述的如式c-3a或式c-3所示化合物消失或不再反应为准。所述的反应的时间优选0.5~3h,例如0.5h、1h、1.5h或3h。
[0020]
在本发明某些实施方案中,步骤(1)中,所述的浓缩可为减压浓缩。
[0021]
在本发明某些实施方案中,步骤(1)中,在所述的浓缩之前,还可进一步包括淬灭步骤,所述的淬灭所用的溶剂可为碱和水。所述的碱可为无机碱,又可为碱金属的碳酸氢盐,例如碳酸氢钠。
[0022]
在本发明某些实施方案中,步骤(2)中,所述的萃取的操作与本领域常规的操作相同。
[0023]
在本发明某些实施方案中,步骤(2)中,在所述的萃取之后,还可包括洗涤操作。所述的洗涤的试剂可为饱和食盐水。
[0024]
在本发明某些实施方案中,步骤(2)中,在所述的萃取之后,还可将所述萃取所得的有机相进行干燥。所述的干燥的试剂可为本领域常规的试剂,例如无水硫酸钠和/或无水硫酸镁。
[0025]
在本发明某些实施方案中,步骤(2)中,在所述的萃取之后,还可将所述萃取所得的有机相进行脱色。所述的脱色的试剂可为本领域常规的试剂,例如活性炭。所述的脱色可在加热条件下进行,所述的加热的温度可为试剂回流的温度。所述的脱色的时间可为0.5-3h,例如1h。
[0026]
在本发明某些实施方案中,步骤(2)中,所述的浓缩结束后,还可进一步包括以下步骤:将浓缩得到的物质与二氯甲烷和正庚烷混合,析出所述的如式c所示化合物。
[0027]
在本发明某些实施方案中,所述的正庚烷和所述的二氯甲烷的体积比优选2:1~50:1,更优选2:1~5:1,例如2:1。
[0028]
在本发明某些实施方案中,所述的二氯甲烷和所述如式c所示化合物的体积质量比优选2~50ml/g。
[0029]
在本发明某些实施方案中,所述的正庚烷的加入方式优选滴加。
[0030]
在本发明某些实施方案中,所述的正庚烷加入时的温度优选10-40℃。
[0031]
在本发明某些实施方案中,所述的正庚烷加入后,搅拌、过滤、干燥,即得所述的如式c所示化合物。所述的搅拌的时间可为0.5-5h,例如0.5h。所述的干燥可为烘干。
[0032]
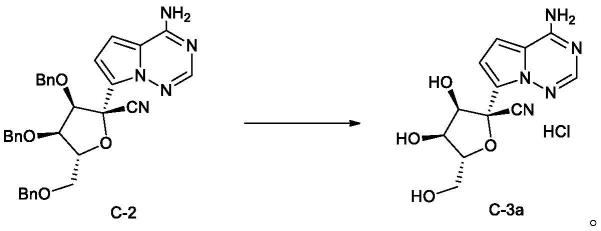
在本发明某些实施方案中,所述的如式c所示化合物的制备方法还可进一步包括以下步骤:有机溶剂中,将如式c-2所示化合物和三氯化硼进行如下所示的脱苄基反应,得所述的如式c-3a所示化合物;
[0033][0034]
在本发明某些实施方案中,所述的有机溶剂可为本领域常规的有机溶剂,优选卤代烃类溶剂和或烷烃类溶剂,例如二氯甲烷和/或正己烷。
[0035]
在本发明某些实施方案中,所述的三氯化硼和所述的如式c-2所示化合物的摩尔比可为本领域常规的摩尔比,优选1:1-5:1,例如1:1、2.7:1或5:1。
[0036]
在本发明某些实施方案中,所述的如式c-2所示化合物和所述的有机溶剂的质量体积比可为本领域常规的质量体积比,优选25~40g/l,例如25g/l、31g/l或40g/l。
[0037]
在本发明某些实施方案中,所述的脱苄基反应的温度可为本领域常规的温度,优选-70~-20℃,例如-40~-20℃或-45℃。
[0038]
在本发明某些实施方案中,所述的脱苄基反应的进程可通过本领域常规的手段进行监控(例如tlc、hplc或lcms),以所述的如式c-2所示化合物消失或不再反应为准。所述的脱苄基反应的时间优选2~3h,例如2h或3h。
[0039]
在本发明某些实施方案中,所述的三氯化硼的加入方式可为本领域常规的加入方式,优选滴加。所述的三氯化硼加入时的温度优选-78~-20℃,例如-78℃、-50℃、-48℃、-47℃、-45℃、-40℃或-20℃。
[0040]
在本发明某些实施方案中,所述的脱苄基反应的后处理可为方法一或方法二,
[0041]
方法一包括以下步骤:将反应液与醇类溶剂混合,分离得所述的如式c-3a所示化合物;
[0042]
方法二包括以下步骤:将反应液与醇类溶剂混合,再将分离得到的固体进行打浆,分离得所述的如式c-3a所示化合物。
[0043]
方法一和方法二中,所述的醇类溶剂可为甲醇、乙醇和异丙醇中的一种或多种,优选甲醇。
[0044]
方法一和方法二中,所述的醇类溶剂和所述的反应液的质量体积比优选40-50g/l(例如43g/l、46g/l或50g/l),更优选为40~46g/l。
[0045]
方法一和方法二中,所述的混合可在-70~25℃下进行,优选在-50~-20℃下进行。
[0046]
方法一和方法二中,所述的混合之后,还可进一步经过搅拌、过滤。所述的搅拌温度优选10-40℃。所述的搅拌的时间优选2~5h,例如2h。所述的过滤优选抽滤。
[0047]
方法二中,所述的打浆的溶剂优选醇类溶剂、或醇类溶剂和酯类溶剂的混合溶剂,例如乙醇、或甲醇和乙酸乙酯的混合溶剂。所述的醇类溶剂优选甲醇和/或乙醇。所述的酯类溶剂优选乙酸乙酯。当所述的溶剂为醇类溶剂和酯类溶剂的混合溶剂时,所述的醇类溶剂和酯类溶剂的体积比优选1:1~1:3,例如1:2。
[0048]
方法二中,所述的打浆的溶剂和所述的固体的体积质量比优选6ml/g~12ml/g(例如6.4ml/g、7ml/g、11.2ml/g或11.4ml/g),更优选6~10ml/g。
[0049]
方法二中,所述的打浆结束后,还可经进一步过滤、干燥,得所述的如式c-3a所示化合物。所述的过滤可为抽滤。所述的干燥可为烘干。所述的干燥的时间可为35~55℃,例如45℃。
[0050]
本发明提供了一种如式c-3a所示化合物的制备方法,其包括以下步骤:有机溶剂中,将如式c-2所示化合物和三氯化硼进行如下所示的脱苄基反应,得所述的如式c-3a所示化合物;
[0051]
[0052]
在本发明某些实施方案中,所述的有机溶剂可为本领域常规的有机溶剂,优选卤代烃类溶剂和或烷烃类溶剂,例如二氯甲烷和/或正己烷。
[0053]
在本发明某些实施方案中,所述的三氯化硼和所述的如式c-2所示化合物的摩尔比可为本领域常规的摩尔比,优选1:1-5:1,例如1:1、2.7:1或5:1。
[0054]
在本发明某些实施方案中,所述的如式c-2所示化合物和所述的有机溶剂的质量体积比可为本领域常规的质量体积比,优选25~40g/l,例如25g/l、31g/l或40g/l。
[0055]
在本发明某些实施方案中,所述的脱苄基反应的温度可为本领域常规的温度,优选-70~-20℃,例如-40~-20℃或-45℃。
[0056]
在本发明某些实施方案中,所述的脱苄基反应的进程可通过本领域常规的手段进行监控(例如tlc、hplc或lcms),以所述的如式c-2所示化合物消失或不再反应为准。所述的脱苄基反应的时间优选2~3h,例如2h或3h。
[0057]
在本发明某些实施方案中,所述的三氯化硼的加入方式可为本领域常规的加入方式,优选滴加。所述的三氯化硼加入时的温度优选-78~-20℃,例如-78℃、-50℃、-48℃、-47℃、-45℃、-40℃或-20℃。
[0058]
在本发明某些实施方案中,所述的脱苄基反应的后处理可为方法一或方法二,
[0059]
方法一包括以下步骤:将反应液与醇类溶剂混合,分离得所述的如式c-3a所示化合物;
[0060]
方法二包括以下步骤:将反应液与醇类溶剂混合,再将分离得到的固体进行打浆,分离得所述的如式c-3a所示化合物。
[0061]
方法一和方法二中,所述的醇类溶剂可为甲醇、乙醇和异丙醇中的一种或多种,优选甲醇。
[0062]
方法一和方法二中,所述的醇类溶剂和所述的反应液的质量体积比优选40-50g/l(例如43g/l、46g/l或50g/l),更优选为40~46g/l。
[0063]
方法一和方法二中,所述的混合可在-70~25℃下进行,优选在-50~-20℃下进行。
[0064]
方法一和方法二中,所述的混合之后,还可进一步经过搅拌、过滤。所述的搅拌温度优选10-40℃。所述的搅拌的时间优选2~5h,例如2h。所述的过滤优选抽滤。
[0065]
方法二中,所述的打浆的溶剂优选醇类溶剂、或醇类溶剂和酯类溶剂的混合溶剂,例如乙醇、或甲醇和乙酸乙酯的混合溶剂。所述的醇类溶剂优选甲醇和/或乙醇。所述的酯类溶剂优选乙酸乙酯。当所述的溶剂为醇类溶剂和酯类溶剂的混合溶剂时,所述的醇类溶剂和酯类溶剂的体积比优选1:1~1:3,例如1:2。
[0066]
方法二中,所述的打浆的溶剂和所述的固体的体积质量比优选6ml/g~12ml/g(例如6.4ml/g、7ml/g、11.2ml/g或11.4ml/g),更优选6~10ml/g。
[0067]
方法二中,所述的打浆结束后,还可经进一步过滤、干燥,得所述的如式c-3a所示化合物。所述的过滤可为抽滤。所述的干燥可为烘干。所述的干燥的时间可为35~55℃,例如45℃。
[0068]
在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
[0069]
本发明所用试剂和原料均市售可得。
[0070]
本发明的积极进步效果在于:
[0071]
(1)化合物c的制备方法中,用二氯甲烷作为萃取溶剂,c-3完全留在水相,未反应完全的c-3a留在水中,减少了杂质残留,便于后续步骤的质量控制。经过重结晶工艺,获得高纯度高收率的化合物c(丙叉物),纯度hplc》99%,收率》80%。避免了文献中报道的柱层析分离,适合工业化大规模生产。
[0072]
(2)化合物c-3a的制备方法中,加入醇类溶剂淬灭反应,析出稳定的c-3a(脱苄物盐酸盐),通过打浆纯化即得到高纯度的c-3a(脱苄物盐酸盐)中间体。在制备中间体c-3a(脱苄物盐酸盐)的过程中,避免了文献报道的在淬灭的过程中需加碱中和,浓缩,反复浸洗以及用甲醇和水进行析晶等繁琐操作,极大的简化了后处理操作方法,得到收率稳定的化合物c-3a(脱苄物盐酸盐)固体,纯度hplc》98%,收率大于等于89%。
具体实施方式
[0073]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0074]
本发明中,由化合物a和b制备化合物c-1、化合物c-1制备化合物c-2,化合物c制备化合物e,化合物e制备化合物f,以及化合物c-3的制备均参照文献nature,2016,531,381-si(2)进行。
[0075]
实施例1:c-3a(脱苄物盐酸盐)的制备
[0076][0077]
在反应瓶中投入c-2(140g)和二氯甲烷(3.5l),降温至-45℃,滴加1m的三氯化硼二氯甲烷溶液(1l),-45℃左右保温反应约2h时反应结束,加入甲醇(160g),升至室温搅拌2h,抽滤,得潮品约100g,hplc纯度98.41%,滤饼用700ml乙醇打浆,抽滤,真空45℃烘干,得到白色固体c-3a(脱苄物盐酸盐)(75g,91%),hplc纯度98.6%。
[0078]1h nmr(d2o)δ8.1(s,1h),7.37(d,j=5.1hz,1h),7.14(d,j=4.8hz,1h),4.94(d,j=5.4hz,1h),4.42(app q,j=4.2hz,1h),4.35(t,j=5.1hz,1h),3.86(dd,j=12.8,3.2hz,1h),3.79(dd,j=12.8,4.7hz,1h)。
[0079]1h nmr(dmso)δ10.1(s,1h),9.3(s,1h),8.2(s,1h),7.49(d,1h),7.0(d,1h),5.7(s,1h),4.5(d,1h),4.0(dd,1h),3.9(t,1h),3.6(dd,1h),3.5(dd,1h)。
[0080]
通过离子色谱(ag11-hc-4μm 4*50guard 4*250mm aralytrcal)测定氯离子含量,c-3a(脱苄物盐酸盐)氯离子含量为13%,即c-3和一分子hcl生成的一分子c-3a(脱苄物盐酸盐)。
[0081]
实施例2:c-3a(脱苄物盐酸盐)的制备
[0082][0083]
在反应瓶中投入c-2(140g)和二氯甲烷(3.5l),降温至-50℃,滴加1m的三氯化硼正己烷溶液,内温控制在-40~-20℃,约25min加完,-45℃左右保温反应,约2h时反应结束,加入甲醇(150g),升至室温搅拌5h,抽滤,得潮品约110g,hplc纯度98.3%滤饼用710ml乙醇打浆,抽滤,真空45℃烘干,得到白色固体c-3a(77g,94%),hplc纯度98.5%。其氢谱数据同实施例1。
[0084]
实施例3:c-3a(脱苄物盐酸盐)的制备
[0085][0086]
在5l反应瓶中投入c-2(140g)和二氯甲烷(3.5l),降温至-47℃,滴加1m的三氯化硼二氯甲烷溶液(1l),内温控制在-40~-20℃,约25min加完,-40~-20℃左右保温反应2-3h,约2h时反应结束,加入甲醇(155g),升至室温搅拌2h,抽滤,得潮品约105g,hplc纯度98.42%滤饼加入甲醇(400ml),乙酸乙酯(800ml)打浆,抽滤,真空45℃烘干,得到白色固体c-3a(73g,89%),hplc纯度98.65%。其氢谱数据同实施例1。
[0087]
实施例4:c-3a(脱苄物盐酸盐)的制备
[0088][0089]
在5l反应瓶中投入c-2(140g)和二氯甲烷(3.5l),降温至-48℃,滴加1m的三氯化硼正己烷溶液(1l),内温控制在-40~-20℃,约25min加完,-40~-20℃左右保温反应2-3h,约2h时反应结束,加入甲醇(148g),升至室温搅拌2-5h,抽滤,得潮品约107g,hplc纯度98.43%滤饼加入甲醇(400ml),乙酸乙酯(800ml)打浆,抽滤,真空45℃烘干,得到白色固体c-3a(74g,90%),hplc纯度98.63%。
[0090]
实施例5:c-3a(脱苄物盐酸盐)的制备
[0091][0092]
在反应瓶中投入c-2(140g)和二氯甲烷(3.2l),降温至-45℃,滴加1m的三氯化硼二氯甲烷溶液(1l),-45℃左右保温反应约2h时反应结束,加入甲醇(160g),升至室温搅拌2h,抽滤,得潮品约100g,hplc纯度98.44%滤饼用700ml乙醇打浆,抽滤,真空45℃烘干,得到白色固体c-3a(脱苄物盐酸盐)(75g,91%),hplc纯度》98.67%。
[0093]
实施例6:c-3a(脱苄物盐酸盐)的制备
[0094][0095]
在反应瓶中投入c-2(140g)和二氯甲烷(3.5l),降温至-45℃,滴加1m的三氯化硼二氯甲烷溶液(1l),-45℃左右保温反应约2h时反应结束,加入甲醇(160g),升至室温搅拌2h,抽滤,得潮品约100g,hplc纯度98.47%滤饼加入甲醇(200ml),乙酸乙酯(400ml)打浆,抽滤,真空45℃烘干,得到白色固体c-3a(脱苄物盐酸盐)(74g,90%),hplc纯度》98.7%。
[0096]
实施例7:c(丙叉物)的制备
[0097][0098]
在100ml反应瓶中投入c-3a(0.62g)和丙酮(16ml),加入2,2-二甲氧基丙烷(1.4ml)和浓硫酸(0.2ml),室温搅拌0.5h后,升温至45℃。反应1h,降温至室温,随后加入固体碳酸氢钠(0.8g)和水(1ml)。混合物搅拌15min后减压浓缩。浓缩得到的粗品加入水(10ml)和二氯甲烷(30ml)。搅拌,静置,分出有机相,水相再用二氯甲烷(10ml)提取。合并有机相,用10ml饱和食盐水洗涤。有机层加入0.2g无水硫酸镁干燥后过滤,滤液浓缩后得到油状物(hplc纯度94.6%,c-3的hplc含量0.18%)。油状物用5ml二氯甲烷溶清,室温下滴加10ml正庚烷,滴加过程中有大量固体析出,滴加完毕后室温搅拌半小时,过滤烘干得到产品c(0.55g,87.7%),hplc纯度99.1%,c-3的hplc含量0.12%。1h nmr(400mhz,dmso)δ8.03-7.84(m,3h),6.91(q,j=4.6hz,2h),5.38(d,j=6.6hz,1h),5.02(t,j=5.7hz,1h),4.90(dd,j=6.6,3.1hz,1h),4.32(td,j=5.3,3.2hz,1h),3.59-3.45(m,2h),1.64(s,3h),1.37
(s,3h)。
[0099]
实施例8:c(丙叉物)的制备
[0100][0101]
在2l反应瓶中投入c-3a(50.8g)和丙酮(1300ml),加入2,2-二甲氧基丙烷(106ml)和浓硫酸(16.4ml),室温搅拌0.5h后,升温至45℃。反应1h,降温至室温,随后加入固体碳酸氢钠(67.5g)和水(100ml)。混合物搅拌15min后减压浓缩。浓缩得到的粗品加入水(400ml)和二氯甲烷(500ml)。搅拌,静置,分出下层有机相,水相再用二氯甲烷(250ml)提取。合并有机相,用250ml饱和食盐水洗涤。有机层加入10g无水硫酸镁,10g活性炭,热回流1小时,热过滤,滤液浓缩后得到油状物(hplc纯度95.4%,c-3的hplc含量0.14%)。油状物用500ml二氯甲烷溶清,室温下滴加1000ml正庚烷,滴加过程中有大量固体析出,滴加完毕后室温搅拌半小时,过滤烘干得到产品c(48g,93.5%),hplc纯度99.5%,c-3的hplc含量0.06%。其氢谱数据同实施例7。
[0102]
实施例9:c(丙叉物)的制备
[0103][0104]
在100ml反应瓶中投入c-3(0.55g)和丙酮(16ml),加入2,2-二甲氧基丙烷(1.4ml)和浓硫酸(0.2ml),室温搅拌0.5h后,升温至45℃。反应1h,降温至室温,随后加入固体碳酸氢钠(0.8g)和水(1ml)。混合物搅拌15min后减压浓缩。浓缩得到的粗品加入水(10ml)和二氯甲烷(30ml)。搅拌,静置,分出下层有机相,水相再用二氯甲烷(10ml)提取。合并有机相,用10ml饱和食盐水洗涤。有机层加入0.2g无水硫酸镁干燥后过滤,滤液浓缩后得到油状物(hplc纯度94.5%,c-3的hplc含量0.18%)。油状物用5ml二氯甲烷溶清,室温下滴加10ml正庚烷,滴加过程中有大量固体析出,滴加完毕后室温搅拌半小时,过滤烘干得到产品c(0.54g,86.3%),hplc纯度99.3%,c-3的hplc含量0.09%。
[0105]
对比例1:c(丙叉物)的制备
[0106][0107]
在100ml反应瓶中投入c-3a(0.62g)和丙酮(16ml),加入2,2-二甲氧基丙烷(1.4ml)和浓硫酸(0.2ml),室温搅拌0.5h后,升温至45℃。反应1h,降温至室温,随后加入固体碳酸氢钠(0.8g)和水(1ml)。混合物搅拌15min后减压浓缩。浓缩得到的粗品加入水(10ml)和乙酸乙酯(30ml)。搅拌,静置,分出有机相,水相再用乙酸乙酯(10ml)提取。合并有机相,用10ml饱和食盐水洗涤。有机层加入0.2g无水硫酸镁干燥后过滤,滤液浓缩后得到油状物(hplc纯度93.8%,c-3的hplc含量1.10%)。油状物用5ml二氯甲烷溶清,室温下滴加10ml正庚烷,滴加过程中有大量固体析出,滴加完毕后室温搅拌半小时,过滤烘干得到产品c(0.55g,87.7%),hplc纯度98.1%,c-3的hplc含量1.1%。
[0108]
对比例2:c(丙叉物)的制备
[0109][0110]
在100ml反应瓶中投入c-3a(0.62g)和丙酮(16ml),加入2,2-二甲氧基丙烷(1.4ml)和浓硫酸(0.2ml),室温搅拌0.5h后,升温至45℃。反应1h,降温至室温,随后加入固体碳酸氢钠(0.8g)和水(1ml)。混合物搅拌15min后减压浓缩。浓缩得到的粗品加入水(10ml)和乙酸乙酯(30ml)。搅拌,静置,分出有机相,水相再用乙酸乙酯(10ml)提取。合并有机相,用10ml饱和食盐水洗涤。有机层加入0.2g无水硫酸镁干燥后过滤,滤液浓缩后得到油状物(hplc纯度94.2%,c-3的hplc含量1.02%)。油状物用4ml乙酸乙酯溶清,室温下滴加8ml正庚烷,滴加过程中有固体析出,滴加完毕后室温搅拌半小时,过滤烘干得到产品c(0.22g,35.1%),hplc纯度98.8%,c-3的hplc含量0.5%。
[0111]
对比例3:c(丙叉物)的制备
[0112][0113]
在100ml反应瓶中投入c-3(0.55g)和丙酮(16ml),加入2,2-二甲氧基丙烷(1.4ml)和浓硫酸(0.2ml),室温搅拌0.5h后,升温至45℃。反应1h,降温至室温,随后加入固体碳酸
氢钠(0.8g)和水(1ml)。混合物搅拌15min后减压浓缩。浓缩得到的粗品加入水(10ml)和乙酸乙酯(30ml)。搅拌,静置,分出有机相,水相再用乙酸乙酯(10ml)提取。合并有机相,用10ml饱和食盐水洗涤。有机层加入0.2g无水硫酸镁干燥后过滤,滤液浓缩后得到油状物(hplc纯度93.3%,c-3的hplc含量1.22%)。油状物用5ml二氯甲烷溶清,室温下滴加10ml正庚烷,滴加过程中有大量固体析出,滴加完毕后室温搅拌半小时,过滤烘干得到产品c(0.54g,86.3%),hplc纯度98.4%,c-3的hplc含量1.1%。
[0114]
对比例4:c(丙叉物)的制备
[0115][0116]
在100ml反应瓶中投入c-3a(0.6g)和丙酮(16ml),加入2,2-二甲氧基丙烷(1.4ml)和浓硫酸(0.2ml),室温搅拌0.5h后,升温至45℃。反应1h,降温至室温,随后加入固体碳酸氢钠(0.8g)和水(1ml)。混合物搅拌15min后减压浓缩。浓缩得到的粗品加入水(10ml),再分别用甲基叔丁基醚、异丙醚、乙酸乙酯和甲苯进行萃取,结果显示,甲基叔丁基醚、异丙醚、乙酸乙酯和甲苯会将产物c和未反应完全的c-3一起萃取出来,后续过程难以进行分离。c的hplc纯度≤95%,c-3的hplc含量1%。
[0117]
对比例5:c(丙叉物)的制备
[0118][0119]
在100ml反应瓶中投入c-3a(0.6g)和丙酮(16ml),加入2,2-二甲氧基丙烷(1.4ml)和浓硫酸(0.2ml),室温搅拌0.5h后,升温至45℃。反应1h,降温至室温,随后加入固体碳酸氢钠(0.8g)和水(1ml)。混合物搅拌15min后减压浓缩。浓缩得到的粗品加入水(10ml)和二氯甲烷(30ml)。搅拌,静置,分出下层有机相,水相再用二氯甲烷(10ml)提取。合并有机相,用10ml饱和食盐水洗涤。有机层加入0.2g无水硫酸镁干燥后过滤,滤液浓缩后得到油状物(hplc纯度94.5%,c-3的hplc含量0.17%)。油状物用3ml甲基叔丁基醚55℃加热溶清,室温下有少量固体析出,过滤烘干得到产品c(0.1g,14.5%),hplc纯度99.3%,c-3的hplc含量0.08%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1