一种简便的特立氟胺制备方法
本发明涉及药物化学技术领域,尤其涉及一种简便的特立氟胺的制备方法。
背景技术:
特立氟胺(teriflunomide),化学名:(2z)-氰基-3-羟基-n-[4-(三氟甲基)苯基]-2-丁烯酰胺。特立氟胺是一种二氢乳清酸脱氢酶(dhodh)抑制剂,是来氟米特的活性代谢产物(leflunomide)。特立氟胺(teriflunomide)是赛诺菲-安万特研制成功的一种新的治疗多发性硬化症(multiplesclerosis,ms)药物,于2012年9月经fda批准上市,适用于治疗成人复发性多发性硬化症。现今国内也对特立氟胺开展深入研究,但主要用于治疗皮肤病。特立氟胺分子式为:c12h9f3n2o2,分子量为270.06,结构如式1。
目前,特立氟胺的制备方法主要归为以下三种:
第一种合成方法:以乙酰乙酸乙酯和原甲酸三乙酯为起始原料,经缩合再与羟胺环合生成5-甲基异噁唑-4-甲酸乙酯,再经水解生成5-甲基异噁唑-4-甲酸,然后与氯化亚砜生成酰氯,再与三氟甲基苯胺缩合制得来氟米特,再将来氟米特在碱性条件下水解开环制得特立氟胺,具体路线如下:
这一种方法反应步骤繁琐,不易于纯化且总收率偏低,且合成过程中使用大量的二氯亚砜,极易腐蚀设备且污染环境。
第二种合成方法:以2-氰基乙酸和对三氟甲基苯胺为起始原料,先将2-氰基乙酸制备成酰氯,然后和对三氟甲基苯胺缩合制得中间体2-氰基-n-(4-三氟甲基苯基)乙酰胺,中间体在强碱作用下与乙酰氯作用制得特立氟胺,具体路线如下:
这种合成方法也需要用到大量的二氯亚砜,对环境伤害较大。
现有的特立氟胺合成方法中,大多数需要用到大量的酰化试剂,反应中产生大量的烟雾及酸,严重腐蚀设备和污染空气,另一方面这些方法合成的中间产物需要纯化,总收率不高不利于工业化生产。
第三种合成方法:以氰基乙酸和4-三氟甲基苯胺为原料,先将氰基乙酸制成活性酯体系,然后与4-三氟甲基苯胺反应得到中间体2-氰基-n-(4-三氟甲基-苯基)-乙酰胺,中间体在强碱作用下与乙酰氯作用制得特立氟胺,具体路线如下:
第三种合成方法虽然没有用到酰化试剂,中间产物也不需要纯化,但是终产物特立氟胺的收率偏低。
技术实现要素:
本发明的目的在于提供一种简便、环保且收率较高的特立氟胺制备方法。
为了实现上述发明目的,本发明提供以下技术方案:
一种简便的特立氟胺的制备方法,包括以下步骤:
(1)将5-甲基异噁唑-4-甲酸和缩合剂在碱性条件下的溶剂中进行缩合反应,得到活性酯体系;
(2)将活性酯体系和4-三氟甲基苯胺在溶剂中进行缩合反应,得到中间体来氟米特;
(3)将得到的中间体来氟米特顺次进行碱处理和酸处理得到特立氟胺。
作为本发明的一种优选的技术方案,步骤(1)中,
所述溶剂为丙酮、乙腈、二氯甲烷、氯仿、四氯化碳、乙酸乙酯、甲苯、苯、二甲苯、n,n-二甲基甲酰胺、二甲基亚砜、乙醚、四氢呋喃和二氧六环中的一种或几种;
所述缩合剂为羰基咪唑、六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷、2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯、二环己基碳二亚胺、1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐、1-羟基-7-氮杂苯并三氮唑、1-羟基苯并三唑、6-氯苯并三氮唑-1,1,3,3-四甲基脲六氟磷酸酯、o-苯并三氮唑-n,n,n',n'-四甲基脲四氟硼酸和氯甲酸异丁酯中的一种或几种;
所述碱性条件所用碱为有机碱或无机碱,所述有机碱为n,n-二甲基甲酰胺、4-二甲氨基吡啶、1,8-二氮杂二环十一碳-7-烯、三乙胺、吡啶、n,n-二异丙基乙胺、n,n-二异丙基乙胺和n-甲基吗啉中的一种或几种,所述无机碱为碳酸钾、碳酸钠、氢氧化钾、氢氧化钠、氢化钠和氢化钾中的一种或几种。
作为本发明的一种优选的技术方案,步骤(1)中,所述5-甲基异噁唑-4-甲酸与缩合剂的摩尔比为1:1.1~5,所述5-甲基异噁唑-4-甲酸与碱的摩尔比为1:1.5~3,所述5-甲基异噁唑-4-甲酸与溶剂的质量体积比为1g:10~30ml。
作为本发明的一种优选的技术方案,步骤(1)中,所述反应体系ph为10~12,反应温度为-5℃~10℃,反应时间为3~5h。
作为本发明的一种优选的技术方案,步骤(2)中,所述溶剂为丙酮、乙腈、二氯甲烷、氯仿、四氯化碳、乙酸乙酯、甲苯、苯、二甲苯、n,n-二甲基甲酰胺、二甲基亚砜、乙醚、四氢呋喃和二氧六环中的一种或几种,所述4-三氟甲基苯胺与溶剂的质量体积比为100g:300ml~700ml。
作为本发明的一种优选的技术方案,所述步骤(1)中的5-甲基异噁唑-4-甲酸与步骤(2)中4-三氟甲基苯胺的摩尔比为1:1.1~3。
作为本发明的一种优选的技术方案,所述步骤(2)中的反应温度为25℃~30℃,反应时间为3~5h。
作为本发明的一种优选的技术方案,步骤(3)中所述碱处理为:将来氟米特和有机碱在溶剂中反应得到中间体a;
所述溶剂为丙酮、乙腈、二氯甲烷、氯仿、四氯化碳、乙酸乙酯、甲苯、苯、二甲苯、n,n-二甲基甲酰胺、二甲基亚砜、乙醚、四氢呋喃和二氧六环中的一种或几种;
所述有机碱为碳酸钾、碳酸钠、氢氧化钾、氢氧化钠、氢化钠和氢化钾中的一种或几种;
所述来氟米特与有机碱的摩尔比为1:3~10,来氟米特与溶剂的质量体积比为1g:10~30ml;
所述碱处理的温度为20~50℃,时间为1~2h。
作为本发明的一种优选的技术方案,步骤(3)中所述酸处理为:将中间体a放入酸溶液中酸化处理;
所述酸溶液为醋酸、稀盐酸和稀硫酸中的一种或几种;
所述酸处理的温度为20~50℃,时间为1~2h。
本发明的有益效果:
本发明提供的特立氟胺的制备方法,反应中间体来氟米特不需纯化,而且操作简便,反应条件温和,产物用水重结晶即可,后处理中废液易于回收,对环境友好,提高了特立氟胺制备的收率。
附图说明
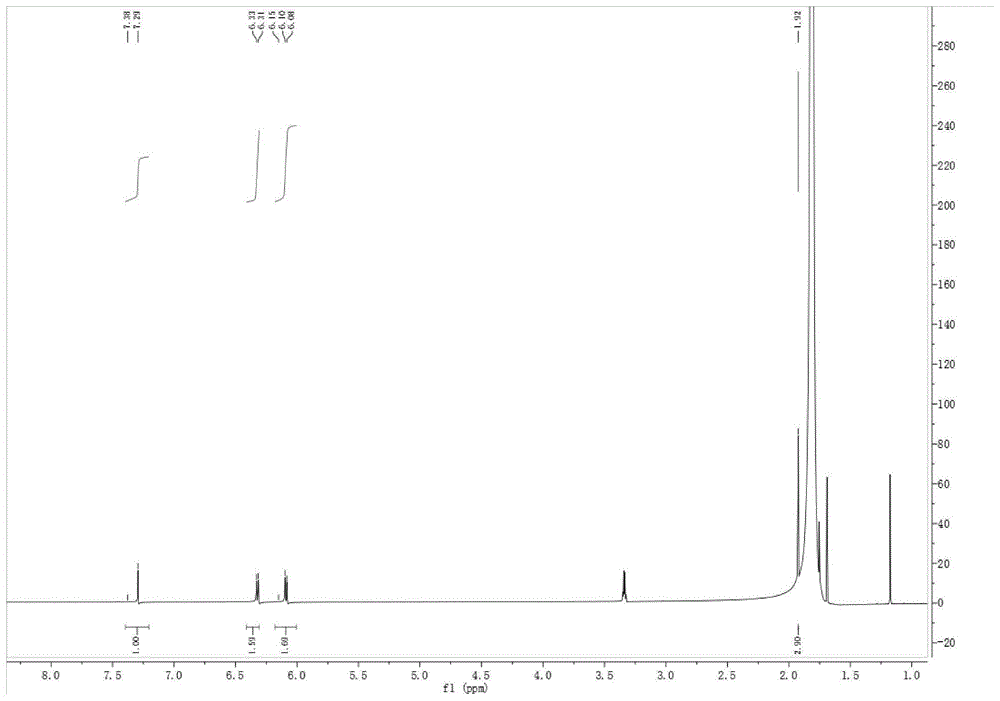
图1为实施例1得到的中间体来氟米特的核磁共振氢谱图。
图2为实施例1得到的产物特立氟胺的核磁共振氢谱图。
具体实施方式
本发明提供了一种简便的特立氟胺的制备方法,包括以下步骤:
(1)将5-甲基异噁唑-4-甲酸和缩合剂在碱性条件下的溶剂中进行缩合反应,得到活性酯体系;
(2)将活性酯体系和4-三氟甲基苯胺在溶剂中进行缩合反应,得到中间体来氟米特;
(3)将得到的中间体来氟米特顺次进行碱处理和酸处理得到特立氟胺。
在本发明步骤(1)中,所述溶剂为丙酮、乙腈、二氯甲烷、氯仿、四氯化碳、乙酸乙酯、甲苯、苯、二甲苯、n,n-二甲基甲酰胺、二甲基亚砜、乙醚、四氢呋喃和二氧六环中的一种或几种,优选为二氯甲烷或n,n-二甲基甲酰胺。
在本发明步骤(1)中,所述缩合剂为羰基咪唑、六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷、2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯、二环己基碳二亚胺、1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐、1-羟基-7-氮杂苯并三氮唑、1-羟基苯并三唑、6-氯苯并三氮唑-1,1,3,3-四甲基脲六氟磷酸酯、o-苯并三氮唑-n,n,n',n'-四甲基脲四氟硼酸和氯甲酸异丁酯中的一种或几种,优选为2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯、六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷、二环己基碳二亚胺和氯甲酸异丁酯中的一种或几种。
在本发明步骤(1)中,所述碱性条件所用碱为有机碱或无机碱,优选为有机碱。所述有机碱为n,n-二甲基甲酰胺、4-二甲氨基吡啶、1,8-二氮杂二环十一碳-7-烯、三乙胺、吡啶、n,n-二异丙基乙胺、n,n-二异丙基乙胺和n-甲基吗啉中的一种或几种,优选为n,n-二异丙基乙胺、三乙胺和n-甲基吗啉中的一种或几种。所述无机碱为碳酸钾、碳酸钠、氢氧化钾、氢氧化钠、氢化钠和氢化钾中的一种或几种,优选为碳酸钠或氢氧化钠。
在本发明步骤(1)中,所述5-甲基异噁唑-4-甲酸与缩合剂的摩尔比为1:1.1~5,优选为1:1.5~2;所述5-甲基异噁唑-4-甲酸与碱的摩尔比为1:1.5~3,优选为1:2~2.5;所述5-甲基异噁唑-4-甲酸与溶剂的质量体积比为1g:10~30ml,优选为1g:10ml。
在本发明中步骤(1)中,所述反应体系ph为10~12,优选为11;所述缩合反应的温度为-5℃~10℃,优选为0℃;所述缩合反应的时间为3~5h,优选为4h。
在本发明步骤(2)中,所述溶剂为丙酮、乙腈、二氯甲烷、氯仿、四氯化碳、乙酸乙酯、甲苯、苯、二甲苯、n,n-二甲基甲酰胺、二甲基亚砜、乙醚、四氢呋喃和二氧六环中的一种或几种,优选为乙酸乙酯。所述4-三氟甲基苯胺与溶剂的质量体积比为100g:300ml~700ml,优选为100g:500ml。
在本发明中,所述步骤(1)中的5-甲基异噁唑-4-甲酸与步骤(2)中4-三氟甲基苯胺的摩尔比为1:1.1~3,优选为1:1.5。
在本发明中,所述步骤(2)中的反应温度为25℃~30℃,优选为28℃,反应时间为3~5h,优选为4h。
在本发明中,步骤(3)中所述碱处理为:将来氟米特和有机碱在溶剂中反应得到中间体a;
所述溶剂为丙酮、乙腈、二氯甲烷、氯仿、四氯化碳、乙酸乙酯、甲苯、苯、二甲苯、n,n-二甲基甲酰胺、二甲基亚砜、乙醚、四氢呋喃和二氧六环中的一种或几种,优选为二氯甲烷或n,n-二甲基甲酰胺。
所述有机碱为碳酸钾、碳酸钠、氢氧化钾、氢氧化钠、氢化钠和氢化钾中的一种或几种,优选为氢氧化钠或氢氧化钾。
所述来氟米特与有机碱的摩尔比为1:3~10,优选为1:3~5,来氟米特与溶剂的质量体积比为1g:10~30ml,优选为1g:10ml。
所述碱处理的温度为20~50℃,优选为35℃,时间为1~2h,优选为1.5h。
在本发明中,步骤(3)中所述酸处理为:将中间体a放入酸溶液中酸化处理;对酸的用量没有具体要求,只要浸没即可。
所述酸溶液为醋酸、稀盐酸和稀硫酸中的一种或几种,优选为稀盐酸。
所述酸处理的温度为20~50℃,优选为35℃,时间为1~2h,优选为1.5h。
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
(1)活性酯的制备
将50g(0.39mol)5-甲基异恶唑-4-甲酸溶于500ml二氯甲烷并置于冰浴;将245g2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(0.65mol)加入上述反应体系中并加入100.81g(0.78mol)n,n-二异丙基乙胺,冰浴搅拌1h后,即得活性酯,此步不需纯化及后处理,直接用于下一步反应。
(2)中间体来氟米特的制备
将100g(0.62mol)4-三氟甲基苯胺加入上述活性酯反应体系中,0℃搅拌3h,tlc跟踪反应至原料反应完毕。反应结束后,将反应液倒入1l冰水中搅拌30min,析出大量白色固体,过滤,滤饼用500ml乙酸乙酯洗涤,烘干称得固体82.5g,收率83%。结构式见于式2。采用核磁共振氢谱对所得白色固体化合物进行鉴定,核磁共振氢谱数据为:1hnmr(600mhz,methanol-d4)δ7.29(s,1h),6.32(d,j=8.5hz,2h),6.09(d,j=8.5hz,2h),1.92(s,3h),谱图见于说明书附图一。
(3)特立氟胺的制备
取中间体来氟米特50g(0.19mol)悬浮于500ml水,加入52.8g(1.32mol)氢氧化钠反应至澄清,接着室温反应2h,tlc跟踪反应至结束。反应结束后,往反应液中滴加浓度1n盐酸,调节ph到2,析出大量乳白色固体,抽滤并烘干固体得到白色固体44.8g,收率90%。结构式见于式3。采用核磁共振氢谱对所得白色固体化合物进行鉴定,核磁共振氢谱数据为:1hnmr(600mhz,dmso-d6)δ10.76(s,1h),7.77(d,j=8.5hz,2h),7.66(d,j=8.6hz,2h),2.26(s,3h),跟所报道文献专利的氢谱位移一致,谱图见于说明书附图二。
实施例2
(1)活性酯的制备
将50g(0.39mol)5-甲基异恶唑-4-甲酸溶于500ml二氯甲烷并置于冰浴;将368.4g(0.65mol)六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷加入上述反应体系中并加入78.93g(0.78mol)三乙胺,冰浴搅拌1h后,即得活性酯,此步不需纯化及后处理,直接用于下一步反应。
(2)中间体来氟米特的制备同实施例1
(3)特立氟胺的制备
取中间体来氟米特50g(0.19mol)悬浮于500ml水,加入74.07g(1.32mol)氢氧化钾反应至澄清,接着室温反应2h,tlc跟踪反应至结束。反应结束后,往反应液中滴加浓度1n盐酸,调节ph到2,析出大量乳白色固体,抽滤并烘干固体得到白色固体47g,收率91%。采用核磁共振氢谱对所得白色固体化合物进行鉴定,经鉴定所得产物的核磁共振氢谱数据和目标产物的氢谱位移一致。
实施例3
(1)活性酯的制备
将50g(0.39mol)5-甲基异恶唑-4-甲酸溶于500ml二氯甲烷并置于冰浴;将88.8g(0.65mol)氯甲酸异丁酯加入上述反应体系中并加入78.90g(0.78mol)n-甲基吗啉,冰浴搅拌1h后,即得活性酯,此步不需纯化及后处理,直接用于下一步反应。
(2)中间体来氟米特的制备同实施例1
(3)特立氟胺的制备
取中间体来氟米特50g(0.19mol)悬浮于500ml水,加入18.2g(1.32mol)氢化钠反应至澄清,接着室温反应2h,tlc跟踪反应至结束。反应结束后,往反应液中滴加浓度1n盐酸,调节ph到2,析出大量乳白色固体,抽滤并烘干固体得到白色固体36g,收率91.5%。采用核磁共振氢谱对所得白色固体化合物进行鉴定,经鉴定所得产物的核磁共振氢谱数据和目标产物的氢谱位移一致。
实施例4
(1)活性酯的制备
将50g(0.39mol)5-甲基异恶唑-4-甲酸溶于500ml二氯甲烷并置于冰浴;将134.1g(0.65mol)二环己基碳二亚胺加入上述反应体系中并加入78.93g(0.78mol)三乙胺,冰浴搅拌1h后,即得活性酯,此步不需纯化及后处理,直接用于下一步反应。
(2)中间体来氟米特的制备同实施例1
(3)特立氟胺的制备
取中间体来氟米特50g(0.19mol)悬浮于500ml水,加入30.3g(1.32mol)氢化钾反应至澄清,接着室温反应2h,tlc跟踪反应至结束。反应结束后,往反应液中滴加浓度1n盐酸,调节ph到2,析出大量乳白色固体,抽滤并烘干固体得到白色固体40g,收率92%。采用核磁共振氢谱对所得白色固体化合物进行鉴定,经鉴定所得产物的核磁共振氢谱数据和目标产物的氢谱位移一致。
实施例5
(1)活性酯的制备
将50g(0.39mol)5-甲基异恶唑-4-甲酸溶于500ml二氯甲烷并置于冰浴;将105.4g(0.65mol)羰基咪唑加入上述反应体系中并加入118.75g(0.78mol)1,8-二氮杂二环十一碳-7-烯,冰浴搅拌1h后,即得活性酯,此步不需纯化及后处理,直接用于下一步反应。
(2)中间体来氟米特的制备同实施例1
(3)特立氟胺的制备
取中间体来氟米特50g(0.19mol)悬浮于500ml水,加入104.5g(1.32mol)碳酸钾反应至澄清,接着室温反应2h,tlc跟踪反应至结束。反应结束后,往反应液中滴加浓度1n盐酸,调节ph到2,析出大量乳白色固体,抽滤并烘干固体得到白色固体66.1g,收率93%。采用核磁共振氢谱对所得白色固体化合物进行鉴定,经鉴定所得产物的核磁共振氢谱数据和目标产物的氢谱位移一致。
实施例6
(1)活性酯的制备
将50g(0.39mol)5-甲基异恶唑-4-甲酸溶于500ml二氯甲烷并置于冰浴;将268.9g(0.65mol)6-氯苯并三氮唑-1,1,3,3-四甲基脲六氟磷酸酯加入上述反应体系中并加入95.30g(0.78mol)4-二甲氨基吡啶,冰浴搅拌1h后,即得活性酯,此步不需纯化及后处理,直接用于下一步反应。
(2)中间体来氟米特的制备同实施例1
(3)特立氟胺的制备
取中间体来氟米特50g(0.19mol)悬浮于500ml水,加入106g(1.32mol)碳酸钠反应至澄清,接着室温反应2h,tlc跟踪反应至结束。反应结束后,往反应液中滴加浓度1n盐酸,调节ph到2,析出大量乳白色固体,抽滤并烘干固体得到白色固体68g,收率95%。采用核磁共振氢谱对所得白色固体化合物进行鉴定,经鉴定所得产物的核磁共振氢谱数据和目标产物的氢谱位移一致。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!