M13噬菌体纳米探针及其制备方法
本发明属于噬菌体纳米探针制备技术领域,具体涉及m13噬菌体纳米探针及其制备方法,尤其涉及m13噬菌体的基因修饰及与金属纳米颗粒组装为m13噬菌体纳米探针及其制备方法。
背景技术:
基于m13噬菌体基因组的可编辑性,其5种外壳蛋白(p3、p6、p8、p7、p9)均可用于展示外源肽或抗体。georgep.smith于1985年首次提出噬菌体展示技术,他成功地将外源dna导入m13中,并凭借噬菌体展示技术获得2018年诺贝尔化学奖。
多年来,m13噬菌体广泛引发了研究者的广泛关注,噬菌体展示技术在免疫学、药理学和药物发现等领域得到了广泛的应用。通过噬菌体展示文库筛选的特异性抗体、肽或其他分子成功表达后,可用于药物递送、疫苗制备、抗病毒治疗、靶向基因治疗、靶向肿瘤治疗和疾病诊断,效果显著。基于m13噬菌体的疾病诊断技术,即多通过噬菌体展示文库筛选获得亲和待检靶标的亲和多肽,或在p3蛋白上展示外源亲和肽序列,然后利用elisa原理及辣根过氧化物酶或链酶亲和生物素放大p8蛋白信号,达到检测待检靶标的目的。这种通过酶与底物的显色反应实现信号放大来实现检测的方法,需要进行多个反应步骤和清洗步骤,操作复杂,灵敏度低,对于病原早期检测及极低浓度检测显示出短板。
m13噬菌体衍生制剂可以在大肠杆菌宿主中复制,降低了生产成本,且高拷贝数的p8蛋白(2700拷贝/pfu)使检测灵敏度更高。但也存在一些不足,如,m13噬菌体高长径和细丝状结构使其在用作检测探针时单分散性差,检测时需要更长的反应时间,且结合效率低;重组m13噬菌体作为病毒能在微生物群落中的大肠杆菌中复制,生物安全性相对较低。
因此,亟需一种安全性高、高效、长度短且检测效率高的噬菌体纳米探针。
技术实现要素:
发明目的:本发明所要解决的技术问题是提供了一种m13噬菌体纳米探针。
本发明还要解决的技术问题是提供了m13噬菌体纳米探针的构建方法。
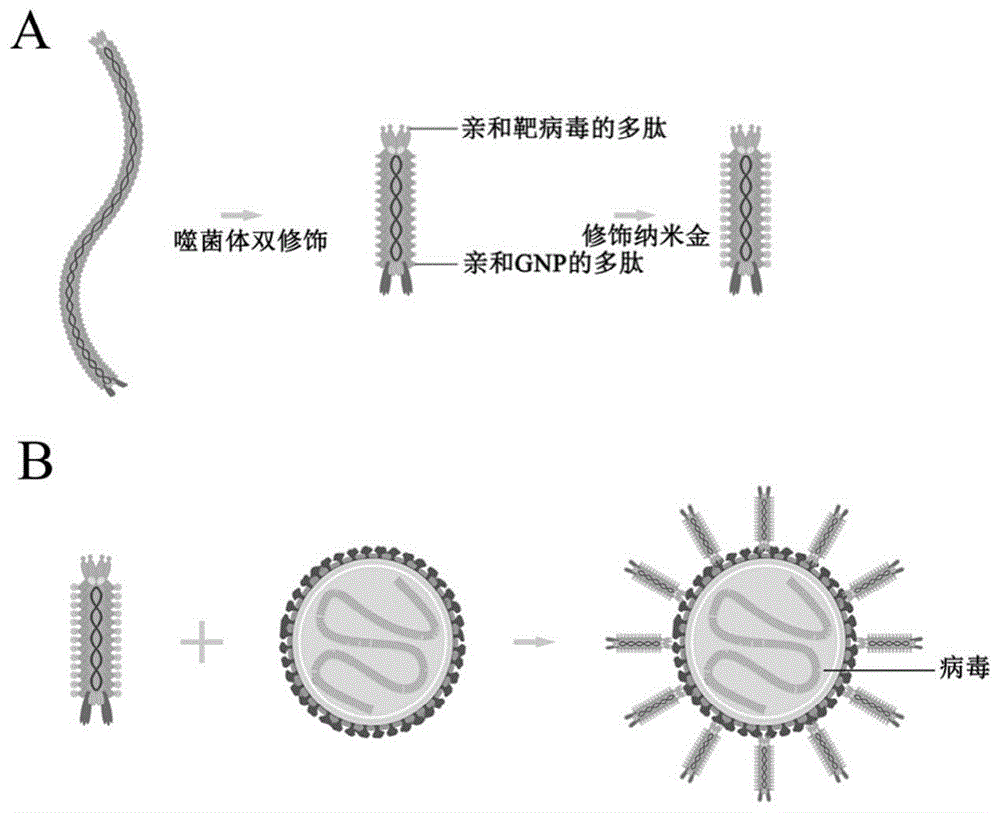
技术方案:为了解决上述技术问题,本发明所采用的技术方案为:一种m13噬菌体纳米探针,所述m13噬菌体纳米探针是先将病毒的亲和多肽展示在辅助噬菌体m13ko7的p3蛋白n端,将纳米粒子的亲和多肽展示在p8蛋白n端得到改造后的辅助噬菌体m13ko7,然后用改造后的辅助噬菌体m13ko7感染含有噬菌粒的菌液,使改造后的辅助噬菌体m13ko7包装噬菌粒,组装成50-100nm的噬菌体纳米探针。
其中,所述病毒包括但不仅限于猪流行性腹泻病毒、流感病毒、新城疫病毒或蓝耳病毒等所有可筛选到亲和肽的病毒,其他病毒、细菌、寄生虫等病原体均可以适用。
其中,所述病毒的亲和多肽是亲和猪流行性腹泻病毒(pedv)的多肽agwyctevlcvq。
其中,所述亲和多肽为agwyctevlcvq、vsgsspds、lkahlppsrlps或ahhahhaad。
其中,所述纳米粒子为金纳米粒子、磁性纳米材料、银纳米材料或量子点,其他纳米粒子均可以适用。
其中,所述金纳米粒子的亲和多肽为-cys-cys-。
其中,所述噬菌粒含有复制起始点ori(+)和包装信号序列片段,所述片段序列如seqidno:7或seqidno:8所示。
作为优选,本发明是通过m13噬菌体的基因修饰并与金纳米颗粒组装为m13噬菌体纳米探针。
本发明内容还包括所述的m13噬菌体纳米探针的构建方法,包括以下步骤:
1)辅助噬菌体m13ko7的p3蛋白n端的改造:利用同源重组将病毒的亲和多肽序列插入至辅助噬菌体m13ko7p3基因序列的n端表达获得阳性克隆;
2)辅助噬菌体m13ko7的p8蛋白n端的改造:在步骤1)获得阳性克隆后利用同源重组将纳米粒子的亲和多肽序列插入至辅助噬菌体m13ko7p8基因序列的n端,筛选获得病毒和纳米粒子的双亲和辅助噬菌体m13ko7;
3)噬菌粒的构建:利用pcr技术扩增m13噬菌体复制起始点ori(+)和包装信号序列片段,所得片段产物经拼接后插入至载体puc57,获得噬菌粒;
4)噬菌体纳米探针制备:将步骤3)获得的噬菌粒转化至大肠杆菌培养得到含有噬菌粒的菌液,加入步骤2)的病毒和纳米粒子的双亲和辅助噬菌体m13ko7,使改造后的辅助噬菌体m13ko7包装噬菌粒,组装成50-100nm的噬菌体纳米探针。
其中,所述步骤1)的辅助噬菌体m13ko7的p3蛋白n端的改造是以m13ko7为扩增模板,通过引物对f3和r1扩增获得pedv亲和多肽碱基序列的片段1,利用引物对f3和r3扩增得到含有pedv亲和多肽碱基序列的片段2,将片段1和片段2经胶回收试剂盒纯化后,用同源重组酶连接,转化得到阳性克隆,所述f3、r3、r1序列分别如seqidno:1、seqidno:2和seqidno:3所示。
其中,所述步骤2)的辅助噬菌体m13ko7的p8蛋白n端的改造是将步骤1)的阳性克隆为模板通过引物对f5和r1扩增出含有亲和金纳米材料多肽碱基序列的片段3,利用引物f2和r5扩增出含有亲和金纳米材料多肽碱基序列的片段4,将片段3和片段4经胶回收试剂盒纯化后,用同源重组酶连接,转化得到阳性克隆,所述r1、f5、f2、r5序列分别如seqidno:3、seqidno:4、seqidno:5和seqidno:6所示。
其中,所述步骤3)的噬菌粒为只含有m13噬菌体的复制起始点和包装信号的质粒,其中,用于制备50nm噬菌体的噬菌粒序列为(seqidno:7):
ccatcgccctgatagacggtttttcgccctttgacgttggagtccacgttctttaatagtggactcttgttccaaactggaacaacactcaaccctatctcgggcaagcttggacgcgccctgtagcggcgcattaagcgcggcgggtgtggtggttacgcgcagcgtgaccgctacacttgccagcgccctagcgcccgctcccgggatcggaattccggccatcgccctgatagacggtttttcgccctttgacgttggagtccacgttctttaatagtggactcttgttccaaactggaaaacgtct,310bp;
其中,用于制备100nm噬菌体的噬菌粒序列为(seqidno:8):
ccatcgccctgatagacggtttttcgccctttgacgttggagtccacgttctttaatagtggactcttgttccaaactggaacaacactcaaccctatctcgggcaagcttggacgcgccctgtagcggcgcattaagcgcggcgggtgtggtggttacgcgcagcgtgaccgctacacttgccagcgccctagcgcccgctcccgggatcggaattccggccatcgccctgatagacggtttttcgccctttgacgttggagtccacgttctttaatagtggactcttgttccaaactggaaaacgtcttacgtagtcgacgttaactcgcgaatgcatctagatccatcgccctgatagacggtttttcgccctttgacgttggagtccacgttctttaatagtggactcttgttccaaactggaacaacactcaaccctatctcgggcaagcttggacgcgccctgtagcggcgcattaagcgcggcgggtgtggtggttacgcgcagcgtgaccgctacacttgccagcgccctagcgcccgctcccgggatcggaattccggccatcgccctgatagacggtttttcgccctttgacgttggagtccacgttctttaatagtggactcttgttccaaactggaaaacgact,656bp。
其中,所述步骤4)的噬菌粒的浓度为od600约等于0.5-0.8时的浓度,即浓度约为2.5×108-4×108cfu/ml。
其中,噬菌体纳米探针长度为50-100nm,直径为6nm。
其中,亲和病毒与纳米粒子的双亲和辅助噬菌体m13ko7,长度为900nm,直径为6nm。
优选地,步骤4)中的噬菌体纳米探针具体制备步骤为:将步骤3)获得的噬菌粒转化至大肠杆菌tg1,待生长至od600约0.5时,加入步骤2)制备的双亲和辅助噬菌体m13ko7并过夜培养,经差异peg/nacl法沉淀后纯化出50-100nm噬菌体探针。
其中,所述亲和金纳米粒子的多肽-cys-cys不仅限于可展示在p8蛋白上,其它外壳蛋白(p3、p6、p7和p9)也可以适用。
本发明利用m13噬菌体展示技术及m13噬菌体的组装原理,制备出可亲和病毒与纳米粒子的纳米噬菌体,其50-100nm的长度相比原长而言,能大大增加探针与待检测病毒的反应概率,因而可大大缩短探针捕获目标的时间;其次,本发明制备的所有噬菌体纳米探针需要借助特定的辅助噬菌体才有完成组装,其自身并不能感染大肠杆菌,亦不能增殖组装成噬菌体;此外,由于金颗粒随着噬菌体纳米探针在待检物表面大量聚积,在临床检测上不需要繁多的清洗和反应步骤也可实现信号放大,实现可视化检测。
有益效果:与现有技术相比,本发明的优点是:
1)本发明利用了m13噬菌体基因组的可编辑性,将亲和病毒的多肽展示在辅助噬菌体m13ko7的p3蛋白n端、亲和纳米粒子的多肽展示在p8蛋白n端;同时利用m13噬菌体的组装原理,改造后的双亲和辅助噬菌体提供主要外壳蛋白,组装噬菌粒成为50-100nm噬菌体。这种纳米噬菌体的复制和组装必须借助辅助噬菌体为其提供外壳蛋白,因此它们保留了与全长噬菌体颗粒相同的外壳蛋白;此外,没有辅助噬菌体参与就无法复制成为纳米粒子,因此噬菌体纳米探针具有很好的生物安全性。
2)本发明引入多肽-cys-cys-序列,使其展示在m13噬菌体p8蛋白n端,成功展示的m13噬菌体经还原剂处理后可打开-cys-cys-形成的二硫键,形成可与金纳米粒子结合的-sh,这种通过-sh与金纳米粒子连接的方式稳定性高于普通亲和多肽直接与金纳米粒子结合,由于-sh与金纳米粒子结合的方式为共价键结合,普通亲和多肽直接与金纳米粒子结合为非共价键结合,共价键的稳定性大于非共价键。
3)本发明的噬菌体纳米探针为m13噬菌体衍生物,长度只有m13噬菌体的1/20-1/10,单分散性好,与待检病原体(如病毒)结合时碰撞效率更高,结合时间更短。
4)本发明的噬菌体纳米探针在与待检病原体(如病毒)结合后可直接加入相应亲和的纳米粒子(如金纳米粒子),实现可视化检测;由于一些纳米粒子(如金纳米粒子、磁纳米粒子)可产生电化学信号,因此可实现基于电化学信号的生物传感器检测探针。
附图说明
图1噬菌体纳米探针制备及检测示意图,a图为金纳米噬菌体的制备过程,b图为制备的金纳米噬菌体与病毒结合的过程;
图2改造后的双亲和辅助噬菌体m13ko7tem表征图;
图3噬菌体纳米探针(50nm)tem表征图;
图4噬菌体纳米探针(100nm)tem表征图;
图5金辅助噬菌体m13ko7与pedv结合的tem表征图;
图6金纳米噬菌体(100nm)与pedv结合的tem表征图;
图7与pedv结合的金辅助噬菌体m13ko7和100nmm13金纳米噬菌体探针数量结果图。
具体实施方式
下面通过具体的实施例对本发明进一步说明,应当指出,对于本领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干变型和改进,这些也应视为属于本发明的保护范围。下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的实验材料,如无特殊说明,均为自常规生化试剂商店购买得到的。
辅助噬菌体m13ko7购自美国neb公司,货号为:n0315s;高保真酶
以下为本发明实施例在pcr和qpcr扩增过程中采用的引物序列、pcr和qpcr扩增体系、以及pcr和qpcr扩增条件。
1、引物
表1应用于本发明的引物
2、高保真酶pcr扩增体系和扩增程序
表2配制总体积为50μl的高保真酶pcr体系
pcr反应程序:98℃30s;95℃10s,50-72℃30s,72℃3min,共计35个循环;72℃7min,4℃50min。
3、菌液pcr扩增体系和扩增程序
表3配制总体积为25μl的菌液pcr反应体系
pcr反应程序:95℃5min;95℃5s,50-72℃30s,72℃45s,共计30个循环;72℃10min,4℃50min。
4、qpcr扩增体系和扩增程序
表4配制总体积为20μl的qpcr反应体系
qpcr反应程序:95℃1min;95℃60s,59℃30s,共计40个循环;59℃30s收集荧光信号。
实施例1:以亲和猪流行性腹泻病毒(pedv)和金纳米粒子探针为例,构建m13ko7噬菌体纳米探针
1、亲和病毒的辅助噬菌体m13ko7改造:以m13ko7为引物设计模版、设计以亲和pedv多肽(agwyctevlcvq)的碱基序列(gcggggtggtattgtacggaggtgttgtgtgttcag)为同源臂的引物,亲和pedv多肽序列插入至p3蛋白的信号肽末端与第一个氨基酸之间(表1,引物f1、r2、f3、r3下划线部分为亲和pedv多肽的碱基序列)。以m13ko7为扩增模版,使用高保真酶
2、亲和纳米粒子的辅助噬菌体m13ko7改造:以步骤1)获得阳性克隆为引物设计模板,设计引物将亲和金纳米材料多肽(-cys-cys-)的碱基序列(tgctgc)插入至p8蛋白n端(即p8蛋白的信号肽末端与第一个氨基酸之间),引物序列见表1(f5和r5,下划线部分为亲和金纳米材料多肽的碱基序列)。以步骤1)获得阳性克隆为扩增模板,使用高保真酶
测序获得的阳性克隆扩大培养过夜,次日4500rpm离心5min,收集上清于新的50ml离心管中,加入上清4倍体积的20%peg8000/2.5mnacl,室温混匀沉淀2h后,12000rpm离心15min,弃上清,1/20体积加入pbs重悬沉淀。重悬物转移至新的ep管中,快速离心去除细胞碎片。取上清到新的ep管中,加入1/5体积的20%peg8000/2.5mnacl,冰浴1h。14000rpm离心10min,弃上清,1/20体积加入pbs重悬沉淀。重悬后的产物即为亲和pedv与金纳米材料的辅助噬菌体,取100μl用于透射电镜表征,表征见图2;剩余部分加入等体积甘油,-20℃保存备用。
3、噬菌粒的构建:
50nm噬菌体探针对应的噬菌粒(序列如seqidno:1所示)交由武汉金开瑞生物科技有限公司合成,并连接至高拷贝载体puc57,转化至大肠杆菌tg1感受态细胞,37℃过夜培养。次日挑取单克隆培养2h,将培养菌液送至北京擎科生物科技有限公司进行测序分析,测序引物为通用t7引物。本步骤高保真酶pcr、菌液pcr的反应体系和反应程序同前所述;得到阳性克隆噬菌粒菌液1。
100nm噬菌体探针对应的噬菌粒由实验室构建(序列如seqidno:2所示),即以制备50nm噬菌体探针对应的噬菌粒为模版设计引物(表1,f7和r7,f8和r8,下划线部分为插入的多克隆位点snabi、sali和hpai),设计引物f7和r7并使用高保真酶
4、噬菌体纳米探针制备:将2种阳性克隆噬菌粒菌液分别进行扩大培养,待生长至od600约0.5时,加入制备的双亲和辅助噬菌体m13ko7(亲和pedv与金纳米材料的辅助噬菌体)并过夜培养,次日4500rpm离心5min,收集上清于新的50ml离心管中,加入终浓度为15%的peg8000/2.5mnacl,室温沉淀4-6h后,12000rpm离心20min,弃上清,1/20体积pbs重悬沉淀,加入终浓度为2.5%的peg8000室温沉淀1h,12000rpm离心10min,分离上清和沉淀。其中,上清分别为50nm和100nmm13噬菌体探针,取100μl用于透射电镜表征,剩余部分加入等体积甘油,-20℃保存备用;沉淀为全长辅助噬菌体,1/20体积pbs重悬沉淀,加入等体积甘油,-20℃保存备用。
其中,50nmm13噬菌体探针tem表征见图3;100nmm13噬菌体探针tem表征见图4。
5、噬菌体修饰金纳米粒子:分别取制备好的双亲和辅助噬菌体m13ko7、50nm的m13噬菌体纳米探针、100nmm13纳米噬菌体探针各10μl,加入90μlpbs缓冲液稀释至100μl,加入终浓度为0.01m的三(2-羧乙基)膦盐酸盐(tcep),室温反应15min,加入200μl直径为5nm的金纳米粒子,室温旋转混匀结合2-4h。7000rpm离心5min去除未结合的金纳米粒子和噬菌体即得金辅助噬菌体m13ko7、50nm的m13金纳米噬菌体纳米探针、100nmm13金纳米噬菌体探针。
6、结合病毒:将经步骤5修饰金纳米粒子后获得的金辅助噬菌体m13ko7和100nmm13金纳米噬菌体探针分别取1013pfu与50μl纯化的pedv混合,室温旋转混匀结合2-4h。7000rpm离心5min去除未结合的金噬菌体探针和pedv。200μl重悬沉淀物为两种pedv@m13@au复合物,取其中50μl用于tem观察,其余150μl用于m13dna提取,并进行qpcr。2×qpcrmastermix、引物f9和r9扩增双亲和辅助噬菌体m13ko7,2×qpcrmastermix、引物f10和r10扩增100nmm13金纳米噬菌体探针。本步骤qpcr的反应体系和反应程序同前所述。
金辅助噬菌体m13ko7与pedv结合的tem表征图见图5;100nm金纳米噬菌体与pedv结合的tem表征图见图6;与pedv结合的金辅助噬菌体m13ko7和100nmm13金纳米噬菌体探针数量结果见图7。
从图5可以看出,首先,由于原长的噬菌体过长,离心不能完全去除未结合的金纳米粒子和噬菌体;其次,在与相同反应时间、相同反应浓度、相同直径的金纳米粒子结合时,由于原长900nm的m13噬菌体太长、分散性较差,导致病毒极大可能不是由于与m13结合被捕获,而是被噬菌体团住而被捕获,这将可能导致非特异性结合的概率增加;最后,图7的qpcr结果显示,两种pedv@m13@au复合物中100nmm13噬菌体数量高于原长m13噬菌体约100倍;因此,原长900nm的m13噬菌体结合效率和捕获效果不如100nm噬菌体。
序列表
<110>扬州大学
<120>m13噬菌体纳米探针及其制备方法
<160>8
<170>siposequencelisting1.0
<210>1
<211>54
<212>dna
<213>人工序列(f3)
<400>1
gcggggtggtattgtacggaggtgttgtgtgttcagggtggaggttcggccgaa54
<210>2
<211>52
<212>dna
<213>人工序列(r3)
<400>2
ctgaacacacaacacctccgtacaataccaccccgcggagtgagaatagaaa52
<210>3
<211>28
<212>dna
<213>人工序列(r1)
<400>3
aagccatttccgctcgccgcagtcgaac28
<210>4
<211>30
<212>dna
<213>人工序列(f5)
<400>4
ccgatgctgtctttcgcttgctgcgctgag30
<210>5
<211>28
<212>dna
<213>人工序列(f2)
<400>5
gttcgactgcggcgagcggaaatggctt28
<210>6
<211>24
<212>dna
<213>人工序列(r5)
<400>6
ggccgcttttgcgggatcgcagca24
<210>7
<211>310
<212>dna
<213>噬菌粒1(artificialsequence)
<400>7
ccatcgccctgatagacggtttttcgccctttgacgttggagtccacgttctttaatagt60
ggactcttgttccaaactggaacaacactcaaccctatctcgggcaagcttggacgcgcc120
ctgtagcggcgcattaagcgcggcgggtgtggtggttacgcgcagcgtgaccgctacact180
tgccagcgccctagcgcccgctcccgggatcggaattccggccatcgccctgatagacgg240
tttttcgccctttgacgttggagtccacgttctttaatagtggactcttgttccaaactg300
gaaaacgtct310
<210>8
<211>656
<212>dna
<213>噬菌粒2(artificialsequence)
<400>8
ccatcgccctgatagacggtttttcgccctttgacgttggagtccacgttctttaatagt60
ggactcttgttccaaactggaacaacactcaaccctatctcgggcaagcttggacgcgcc120
ctgtagcggcgcattaagcgcggcgggtgtggtggttacgcgcagcgtgaccgctacact180
tgccagcgccctagcgcccgctcccgggatcggaattccggccatcgccctgatagacgg240
tttttcgccctttgacgttggagtccacgttctttaatagtggactcttgttccaaactg300
gaaaacgtcttacgtagtcgacgttaactcgcgaatgcatctagatccatcgccctgata360
gacggtttttcgccctttgacgttggagtccacgttctttaatagtggactcttgttcca420
aactggaacaacactcaaccctatctcgggcaagcttggacgcgccctgtagcggcgcat480
taagcgcggcgggtgtggtggttacgcgcagcgtgaccgctacacttgccagcgccctag540
cgcccgctcccgggatcggaattccggccatcgccctgatagacggtttttcgccctttg600
acgttggagtccacgttctttaatagtggactcttgttccaaactggaaaacgact656
- 还没有人留言评论。精彩留言会获得点赞!