一种香橼果实多糖及其制备方法与应用
1.本发明属于中药领域,具体涉及一种香橼果实多糖及其制备方法与应用。
背景技术:
2.多糖是由大于10个醛糖或酮糖脱水缩合而成的结构多样的高分子聚合物,广泛存在于几乎所有的生物体中,尤其是植物细胞壁中。天然来源的植物多糖被称作生物反应调节剂,具有多种药理活性如抗氧化,抗癌,降糖,胃肠保护,抗骨质疏松,抗菌等。
3.香橼(citrus medica l.)是芸香科(rutaceae)柑橘属(citrus)药食同源品种之一,性温,味苦而略甜,归肺、肝、脾经,具有降痰宽中、疏肝理气等功效。我国民间多用于治疗呕吐、咳嗽、消化不良等。香橼化学成分丰富,目前其多酚类、精油、天然色素等已有研究,但对于生理活性广且毒副作用小的多糖成分未有涉及。香橼作为药食两用的天然保健品,其有效成分的提取纯化、药理活性等方面仍需进一步研究,这对于加强香橼的生物利用、开发新药、指导临床用药等具有重要意义。
技术实现要素:
4.为了对多糖结构有更加深刻的研究,本发明的首要目的在于提供一种香橼果实多糖及其制备方法。本发明的香橼果实多糖为两种香橼果实精制多糖,分别命名为cm
‑
1和cm
‑
2,两种精制多糖与现有的多糖不同,其中,cm
‑
1为一种阿拉伯木聚糖,以1,4
‑
β
‑
d
‑
xylp为主链,在o
‑
3位被α
‑
l
‑
araf单取代;cm
‑
1主要由阿拉伯糖,木糖,甘露糖和葡萄糖组成,摩尔比为(10.5~11)∶(11.2~12)∶1∶(1.5~2)。cm
‑
2为一种半乳阿拉伯聚糖,以(1
→
5)
‑
α
‑
l
‑
araf为主链,在o
‑
2或/和o
‑
3位置主要被β
‑
d
‑
galp取代,cm
‑
2主要由阿拉伯糖,甘露糖,葡萄糖和半乳糖组成,摩尔比为(25~26)∶(1~2)∶1∶(6~7)。
5.本发明的另一目的在于提供上述香橼果实多糖在制备降糖、抗皮肤光老化、免疫增强药物中的应用。
6.本发明的目的通过下述方案实现:
7.一种香橼果实多糖的制备方法,包含如下步骤:
8.1)制备香橼(citrus medica l.)果实水提取物,脱色,脱蛋白,醇沉,将沉淀物干燥,得到香橼果实粗多糖(cm);
9.2)将香橼果实粗多糖(cm)经过阴离子交换柱的分离纯化,洗脱剂依次为水和氯化钠溶液,获得两种洗脱组分;分别将两种洗脱组分经过凝胶柱的分离纯化,洗脱剂为水,获得两种精制多糖,分别记为香橼果实精制多糖cm
‑
1和cm
‑
2。
10.步骤1)中所述香橼果实粗多糖的具体制备步骤为
11.s1、将香橼(citrus medica l.)果实粗粉采用有机溶剂进行脱脂,获得脱脂的香橼(citrus medica l.)果实粉;
12.s2、将脱脂的香橼果实粉进行热水浸提,获得香橼果实水提取物;脱色,脱蛋白,醇沉,将沉淀物干燥,获得香橼果实粗多糖。
13.步骤s1中所述有机溶剂为无水乙醇和石油醚;分别采用无水乙醇和石油醚浸泡香橼果实粗粉;分别浸泡的条件为于20~30℃浸泡4~6h;
14.步骤s1中所述香橼果实粗粉是指将香橼果实进行干燥,粉碎过筛;干燥温度为45~60℃;所述粉碎过筛为30~55目筛。
15.步骤s2中所述热水浸提的条件:料液比1∶(10~30)(g/ml),提取的温度为70~95℃,提取的次数为2~6次,单次提取的时间为60~150min。
16.步骤s2中热水浸提后,合并提取液,过滤,滤液浓缩,得香橼果实水提取物。所述浓缩为40~60℃减压浓缩。
17.步骤s2中脱色:将香橼果实水提物与脱色树脂混合,进行脱色处理;抽滤,滤液浓缩;所述浓缩均为40~60℃减压浓缩;
18.所述脱色树脂为d354fd树脂和/或阴离子交换树脂a
‑
722mp。
19.所述树脂在使用前进行预处理;所述预处理的方法为:将树脂采用水浸泡2~5h。浸泡的温度为20~25℃。
20.所述脱色处理的条件为于20~55℃处理2~5h。所述脱色处理在搅拌的条件下进行,搅拌转速10~20r/min。
21.脱蛋白:使用sevage试剂对脱色后的糖液进行脱蛋白处理;收集脱蛋白糖液并浓缩;sevage试剂:正丁醇∶氯仿=1∶(3~5),v/v。所述浓缩为40~65℃旋转浓缩。
22.所述脱蛋白是将sevage试剂加至3~5倍体积的糖液中;常温重复3~6次。
23.所述醇沉是指向脱蛋白的糖液中加入沉淀剂进行沉淀,离心后弃去上清,收集沉淀物。
24.所述醇沉的温度0~4℃;醇沉的时间15~24h;
25.步骤s2中所述的沉淀剂为无水乙醇或体积分数为85~95%乙醇;加入沉淀剂体积是脱蛋白糖液的3~5倍;
26.醇沉时,离心的转速为3500~5500r/min;所述离心的时间为9~24min。
27.步骤s2中冷冻干燥条件:用水溶解沉淀物,置于
‑
20℃冰箱预冻至固态;置于冷冻干燥机真空冻干48~72h。
28.步骤2)中所述阴离子交换柱为阴离子交换柱deae sepharose fast flow;所述凝胶柱为丙烯葡聚糖凝胶sephacryl s 100hr和/或交联葡聚糖凝胶sephadex g
‑
100。
29.步骤2)中将香橼果实粗多糖(cm)经过阴离子交换柱的分离纯化时,依次采用水和氯化钠溶液进行洗脱;氯化钠溶液的浓度为0.01~0.1mol/l,优选为0.05mol/l;洗脱时,流速为1~2ml/min。
30.所述经过凝胶柱的分离纯化,洗脱时流速为0.3~0.6ml/min。
31.步骤2)的具体步骤:
32.p1:阴离子交换柱中装柱:将经预处理的deae sepharose fast flow脱气;装入层析柱;平衡进出口流速;
33.p2:阴离子交换柱中上样:将香橼果实粗多糖(cm)采用水溶解,滤膜过滤,上样,静置;
34.p3:阴离子交换柱中洗脱:依次用水,nacl溶液洗脱,流速为1~2ml/min,每管收集2~5ml,每种洗脱剂收集50~100管;苯酚硫酸法跟踪检测,使用紫外分光光度计于490nm处
测定吸光值;分别以管数、od 490nm作为横、纵坐标,绘制洗脱曲线;分别合并不同洗脱剂对应的洗脱液;减压浓缩;透析,干燥,获得两种洗脱组分记为cm
‑
fr.1和cm
‑
fr.2;所述透析是指截留量3~8kda的透析袋中透析;
35.p4:凝胶柱中装柱:将凝胶脱气装入层析柱;平衡进出口流速;
36.p5:凝胶柱中上样:将阴离子交换柱中所得到的两种洗脱组分分别采用水溶解,滤膜过滤,上样,静置;
37.p6:凝胶柱中洗脱:用水洗脱,流速为0.3~0.6ml/min,每管收集1~2ml,每种洗脱剂收集40~60管;苯酚硫酸法跟踪检测,使用紫外分光光度计于490nm处测定吸光值;分别以管数、od 490nm作为横、纵坐标,绘制洗脱曲线;分别合并cm
‑
fr.1和cm
‑
fr.2经过洗脱的洗脱液;减压浓缩;干燥,获得两种精制多糖。
38.所述deae sepharose fast flow的预处理方式为:先于20~25℃水浸泡12~48h,间歇搅拌;再使用质量浓度为3%~5%稀盐酸浸泡30~60min,用水反复冲洗至酸度计测得溶液ph=7;使用质量浓度为3%~5%naoh溶液浸泡30~60min,去离子水反复冲洗至酸度计测得溶液ph=8~9;
39.所述凝胶的预处理方式为:凝胶为sephacryl s
‑
100hr时,将sephacryl s
‑
100hr于25~35℃用水浸泡20~30h,10~20r/min磁力搅拌2~3h;凝胶为sephadex g
‑
100时,用水于80~100℃浸泡sephadex g
‑
1002~4h;10~20r/min磁力搅拌40~60min;置于20~25℃冷却。
40.凝胶为sephacryl s
‑
100hr和sephadex g
‑
100时,多糖依次经过sephacryl s
‑
100hr凝胶柱和sephadex g
‑
100凝胶柱纯化。
41.步骤p1和p4中所述脱气是指超声除气泡;步骤p2和p5中所述滤膜的孔径为0.22~0.45μm。步骤p3中所述透析条件:温度0~4℃,时间24~72h。
42.步骤p6中所述干燥为冷冻干燥。置于
‑
20℃冰箱预冻至固态;置于冷冻干燥机真空冻干48~72h。
43.所述香橼果实多糖为上述香橼果实精制多糖中一种以上。本发明的香橼果实精制多糖,分别命名为cm
‑
1和cm
‑
2,两种精制多糖与现有的多糖不同,其中,cm
‑
1为一种阿拉伯木聚糖,以1,4
‑
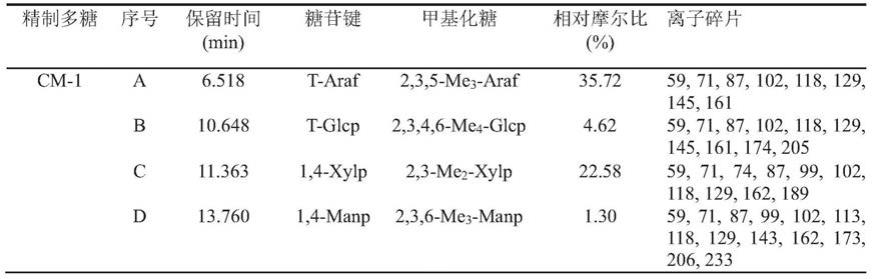
β
‑
d
‑
xylp为主链,在o
‑
3位被α
‑
l
‑
araf单取代;cm
‑
1主要由阿拉伯糖,木糖,甘露糖和葡萄糖组成,摩尔比为(10.5~11)∶(11.2~12)∶1∶(1.5~2),优选为(10.7~10.8)∶(11.5~11.6)∶1∶(1.6~1.8),更优选为10.78∶11.53∶1.00∶1.70。所述香橼果实精制多糖cm
‑
1分子量相对均一,重均平均分子量(mw)为21520da,多分散性指数(pdi)为1.227493。
44.cm
‑
2为一种半乳阿拉伯聚糖,以(1
→
5)
‑
α
‑
l
‑
araf为主链,在o
‑
2或/和o
‑
3位置主要被β
‑
d
‑
galp取代,cm
‑
2主要由阿拉伯糖,甘露糖,葡萄糖和半乳糖组成,摩尔比为(25~26)∶(1~2)∶1∶(6~7),优选为(25.4~25.6)∶(1.4~1.5)∶1∶(6.5~6.6),更优选为25.46∶1.45∶1.00∶6.57。所述香橼果实精制多糖cm
‑
2分子量相对均一,重均平均分子量(mw)为22303da,多分散性指数(pdi)为1.219320。
45.所述香橼果实精制多糖cm
‑
1和cm
‑
2在制备降血糖药物、抗皮肤光老化药物和/或免疫增强药物中的应用。
46.所述香橼果实精制多糖用于制备抑制α
‑
葡萄糖苷酶和/或α
‑
淀粉酶的抑制剂。
47.所述香橼果实精制多糖用于制备修复光损伤皮肤的制剂。
48.一种降糖、抗皮肤光老化、免疫增强药物,含有上述香橼果实精制多糖cm
‑
1和cm
‑
2的至少一种。
49.本发明的原理:本发明在体外细胞水平探究来源于药食两用的香橼果实精制多糖的药理活性,如降糖、抗皮肤光老化、免疫增强等;具有样品用量低、毒副作用小等优点。
50.本发明相较于现有技术具有如下优点及药用价值:
51.(1)本发明的香橼果实精制多糖cm
‑
1和cm
‑
2对α
‑
淀粉酶和α
‑
葡萄糖苷酶具有抑制作用,可以显著提高胰岛素抵抗
‑
肝癌细胞(hepg2
‑
ir)葡萄糖消耗量;
52.(2)本发明的香橼果实精制多糖cm
‑
1和cm
‑
2对uvb损伤的hacat细胞具有显著的修复效果;
53.(3)本发明发现香橼果实精制多糖cm
‑
1和cm
‑
2可以激活小鼠巨噬细胞raw264.7;
54.(4)本发明的香橼果实精制多糖cm
‑
1和cm
‑
2可以增强小鼠巨噬细胞raw264.7的胞饮能力;
55.(5)本发明的香橼果实精制多糖cm
‑
1和cm
‑
2可以诱导小鼠巨噬细胞raw264.7释放tnf
‑
α,il
‑
6和no;
56.(6)本发明的香橼果实精制多糖cm
‑
1和cm
‑
2可以上调小鼠巨噬细胞raw264.7的tnf
‑
α,il
‑
6和inos mrna的表达;
57.(7)本发明的香橼果实精制多糖cm
‑
1和cm
‑
2用药剂量低,且毒副作用小。
附图说明
58.图1是实施例4制备得到的cm
‑
fr.1和cm
‑
fr.2的deae sepharose fast flow阴离子交换柱洗脱曲线图;
59.图2是实施例4得到的cm
‑
1和cm
‑
2的sephadex g
‑
100凝胶柱洗脱曲线图;其中a为cm
‑
1的sephadex g
‑
100凝胶柱洗脱曲线图,b为cm
‑
2的sephadex g
‑
100凝胶柱洗脱曲线图;
60.图3是实施例4得到的cm
‑
1和cm
‑
2的高效凝胶渗透色谱图(hpgpc);其中a为cm
‑
1的hpgpc图,b为cm
‑
2的hpgpc图;
61.图4是实施例4得到的cm
‑
1和cm
‑
2傅里叶红外光谱图(ft
‑
ir);其中a为cm
‑
1的ft
‑
ir图,b为cm
‑
2的ft
‑
ir图;
62.图5是实施例4得到的cm
‑
1的核磁共振图谱(nmr)和化学结构;a为1h nmr,b为
13
c nmr,c为hsqc,d为cosy,e为hmbc,f为化学结构;
63.图6是实施例4得到的cm
‑
2的核磁共振图谱(nmr)和化学结构;a为1h nmr,b为
13
c nmr,c为hsqc,d为cosy,e为hmbc,f为化学结构;
64.图7是实施例4得到的cm
‑
1和cm
‑
2扫描电镜图(sem);其中a为cm
‑
1的sem图,b为cm
‑
2的sem图;
65.图8是实施例4得到的cm
‑
1和cm
‑
2的热稳定性分析图(tg
‑
dsc);其中a为cm
‑
1的tg
‑
dsc图,b为cm
‑
2的tg
‑
dsc图;
66.图9是实施例4得到的cm
‑
1和cm
‑
2对α
‑
淀粉酶抑制率的曲线图;
67.图10是实施例4得到的cm
‑
1和cm
‑
2对α
‑
葡萄糖苷酶抑制率的曲线图;
68.图11是实施例4得到的cm
‑
1和cm
‑
2对胰岛素抵抗
‑
肝癌细胞(hepg2
‑
ir)葡萄糖消
耗量的影响;a是cm
‑
1和cm
‑
2对正常hepg2细胞存活率影响的柱形图;b是cm
‑
1和cm
‑
2对hepg2
‑
ir细胞存活率影响的柱型图;c是cm
‑
1和cm
‑
2对hepg2
‑
ir细胞葡萄糖消耗量影响的柱形图;
69.图12是实施例4得到的cm
‑
1和cm
‑
2对uvb损伤的hacat细胞修复的影响以及对正常的hacat细胞存活率影响;a是cm
‑
1和cm
‑
2对正常的hacat细胞存活率影响的柱形图;b是cm
‑
1和cm
‑
2对uvb损伤的hacat细胞修复影响的柱形图;
70.图13是实施例4得到的cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7的激活作用;a是cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7存活率影响的柱形图;b是cm
‑
1和cm
‑
2激活小鼠巨噬细胞raw264.7倒置显微镜拍照图;
71.图14是实施例4得到的cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7吞噬中性红影响的柱形图;
72.图15是实施例4得到的cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7释放tnf
‑
α,il
‑
6和no影响的柱形图;a是cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7释放tnf
‑
α影响的柱形图;b是cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7释放il
‑
6影响的柱形图;c是cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7释放no影响的柱形图;
73.图16是实施例4得到的cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7的tnf
‑
α,il
‑
6和inos mrna表达影响的柱形图;a是cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7的tnf
‑
αmrna表达量影响的柱形图;b是cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7的il
‑
6mrna表达量影响的柱形图;c是cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7的inos mrna表达量影响的柱形图。
具体实施方式
74.下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。实施例中,人肝癌细胞hepg2,人永生化表皮细胞hacat,小鼠巨噬细胞raw264.7购自中科院上海生命科学研究院细胞资源中心;香橼果实购自广州清平药材市场。
75.实施例1从香橼果实中制备得到香橼果实粗多糖(cm)
76.(1)香橼(citrus medica l.)果实于55℃恒温烘箱干燥至恒重,粉碎过50目筛,得香橼果实粗粉;将香橼果实粗粉依次采用无水乙醇和石油醚于25℃浸泡5h;然后以料液比1∶25(g/ml),提取温度90℃,单次提取时间120min,提取次数3次热水浸提香橼果实粗粉,取上清液于50℃减压浓缩,得到香橼果实水提物浓缩液;
77.(2)将阴离子交换树脂d354fd于25℃在蒸馏水浸泡4h,获得预处理的d354fd;
78.(3)将步骤(1)浓缩糖液与(2)预处理的d354fd按照1∶1(v∶v)混匀后于55℃,转速10r/min,磁力搅拌4h;过滤,于50℃减压浓缩,得到脱色糖液,;
79.(4)将200ml的脱色浓缩糖液与1000ml sevage试剂(正丁醇∶氯仿=1∶4,v/v)于25℃,转速20r/min,磁力搅拌30min,转入3l分液漏斗脱蛋白3次至中间乳白层消失;取上层清液于60℃减压浓缩;
80.(5)向200ml的脱蛋白浓缩糖液中加入800ml无水乙醇于4℃沉淀20h,间歇搅拌;然后于5000r/min离心10min;取沉淀物于真空条件下冷冻干燥48h,得到香橼果实粗多糖(cm)。
81.实施例2从香橼果实中制备得到香橼果实粗多糖(cm)
82.(1)香橼(citrus medica l.)果实于45℃恒温烘箱干燥至恒重,粉碎过40目筛,得香橼果实粗粉;将香橼果实粗粉依次采用无水乙醇和石油醚于23℃浸泡5.5h;以料液比1∶15(g/ml),提取温度85℃,单次提取时间85min,提取次数5次热水浸提香橼果实粗粉,取上清液于45℃减压浓缩,得到香橼果实水提物浓缩液;
83.(2)将大孔阴离子交换树脂d354fd于23℃在蒸馏水浸泡4.5h,获得预处理的d354fd;
84.(3)将步骤(1)浓缩糖液与(2)预处理的d354fd按照1∶1(v∶v)混匀后于23℃,转速20r/min,磁力搅拌2h;真空抽滤,于45℃减压浓缩,获得脱色浓缩糖液;
85.(4)将200ml脱色浓缩糖液与900ml sevage试剂(正丁醇∶氯仿=1∶3,v/v)于20℃,转速10r/min,磁力搅拌60min,转入2l分液漏斗脱蛋白4次至中间乳白层消失;取上层清液于45℃减压浓缩;
86.(5)向200ml脱蛋白浓缩糖液中加入900ml无水乙醇于0℃沉淀18h,间歇搅拌;然后于5500r/min离心12min;取沉淀物于真空条件下冷冻干燥60h,得到香橼果实粗多糖(cm)。
87.实施例3从香橼果实中制备得到香橼果实粗多糖(cm)
88.(1)香橼(citrus medica l.)果实于50℃恒温烘箱干燥至恒重,粉碎过30目筛,得香橼果实粗粉;将香橼果实粗粉依次采用无水乙醇和石油醚于20℃浸泡6h;以料液比1∶30(g/ml),提取温度93℃,单次提取时间80min,提取次数4次热水浸提香橼果实粗粉,取上清液于55℃减压浓缩,得到香橼果实水提物浓缩液;
89.(2)将大孔弱碱性阴离子交换树脂d354fd于20℃在蒸馏水浸泡5h,获得预处理的d354fd;
90.(3)将步骤(1)浓缩糖液与(2)预处理的d354fd按照1∶1(v∶v)混匀后于25℃,转速15r/min,磁力搅拌2.5h;真空抽滤,于50℃减压浓缩,得到脱色浓缩糖液;
91.(4)将200ml脱色浓缩糖液与800ml sevage试剂(正丁醇∶氯仿=1∶5,v/v)于20℃,转速15r/min,磁力搅拌45min,转入2l分液漏斗脱蛋白5次至中间乳白层消失;取上层清液于55℃减压浓缩;
92.(5)向200ml脱蛋白浓缩糖液中加入1000ml无水乙醇于0℃沉淀22h,间歇搅拌;然后于4500r/min离心15min;取沉淀物于真空条件下冷冻干燥72h,得到香橼果实粗多糖(cm)。
93.实施例4从香橼果实粗多糖(cm)制备精制多糖cm
‑
1和cm
‑294.(1)deae sepharose fast flow预处理:称取deae sepharose fast flow 80g于20℃去离子水浸泡12h,间歇搅拌;再使用5wt%稀盐酸浸泡60min,去离子水反复冲洗至酸度计测得溶液ph=7;使用5wt%naoh溶液浸泡60min,去离子水反复冲洗至酸度计测得溶液ph=8~9;超声除气泡;
95.(2)将(1)预处理的deae sepharose fast flow置于层析柱;平衡进出口流速;
96.(3)使用去离子水充分溶解实施例1得到的香橼果实粗多糖(cm);多糖溶液经0.22μm微孔滤膜过滤,上样,静置8h;
97.(4)依次使用去离子水,0.05mol/l的nacl溶液洗脱,流速为1.5ml/min,每管收集4.5ml,每种洗脱剂收集100管;苯酚硫酸法跟踪检测,使用紫外分光光度计于490nm处测定吸光值;分别以管数、od 490nm作为横、纵坐标,绘制洗脱曲线;分别合并不同洗脱剂对应的
洗脱液;减压浓缩,将浓缩糖液置于透析袋(截留量=8kda)于4℃蒸馏水中透析50h;于真空条件下冷冻干燥48h得到cm
‑
fr.1和cm
‑
fr.2;
98.(5)葡聚糖凝胶sephadex g
‑
100预处理:使用去离子水于100℃沸水浴中浸泡sephadex g
‑
100 2h;15r/min磁力搅拌50min;置于25℃冷却;
99.(6)将经预处理的sephadex g
‑
100凝胶脱气;装入层析柱;恒流泵平衡进出口流速;
100.(7)使用蒸馏水充分溶解cm
‑
fr.1和cm
‑
fr.2;多糖溶液经0.45μm微孔滤膜过滤;分别上样;静置7h;
101.(8)使用蒸馏水洗脱,流速为0.4ml/min,每管收集1.2ml,每种洗脱剂收集50管;苯酚硫酸法跟踪检测,使用紫外分光光度计于490nm处测定吸光值;分别以管数、od 490nm作为横、纵坐标,绘制洗脱曲线;分别合并cm
‑
fr.1和cm
‑
fr.2洗脱过程中对应的洗脱液;减压浓缩;于真空条件下冷冻干燥40h,得香橼果实精制多糖cm
‑
1和cm
‑
2。
102.图1是实施例4制备得到的cm
‑
fr.1和cm
‑
fr.2的deae sepharose fast flow阴离子交换柱洗脱曲线图;
103.图2是实施例4得到的cm
‑
1和cm
‑
2的sephadex g
‑
100凝胶柱洗脱曲线图;其中a为cm
‑
1的sephadex g
‑
100凝胶柱洗脱曲线图,b为cm
‑
2的sephadex g
‑
100凝胶柱洗脱曲线图。
104.实施例5从香橼果实粗多糖(cm)制备精制多糖cm
‑
1和cm
‑2105.(1)deae sepharose fast flow预处理:称取deae sepharose fast flow 80g于25℃去离子水浸泡24h,间歇搅拌;再使用4%稀盐酸浸泡40min,去离子水反复冲洗至酸度计测得溶液ph=7;使用4%naoh溶液浸泡40min,去离子水反复冲洗至酸度计测得溶液ph=8~9;超声除气泡;
106.(2)将(1)预处理的deae sepharose fast flow装入层析柱;恒流泵平衡进出口流速;
107.(3)使用去离子水充分溶解实施例2得到的香橼果实粗多糖(cm)并配置成浓度为11mg/ml的多糖溶液;多糖溶液经0.22μm微孔滤膜过滤;上样;静置8h;
108.(4)依次使用去离子水,0.05mol/l的nacl溶液洗脱,流速为1.3ml/min,每管收集5ml,每种洗脱剂收集50管;苯酚硫酸法跟踪检测,使用紫外分光光度计于490nm处测定吸光值;分别以管数、od 490nm作为横、纵坐标,绘制洗脱曲线;分别合并不同洗脱剂对应的洗脱液;于55℃减压浓缩;将浓缩糖液置于透析袋(截留量=3kda)于0℃蒸馏水中透析72h;于真空条件下冷冻干燥40h得到cm
‑
fr.1和cm
‑
fr.2;
109.(5)交联葡聚糖凝胶sephadex g
‑
100预处理:使用去离子水于90℃浸泡sephadex g
‑
100 3h;10r/min磁力搅拌60min;置于22℃冷却;
110.(6)将经预处理的sephadex g
‑
100凝胶脱气;装入层析柱;恒流泵平衡进出口流速;
111.(7)用去离子水充分溶解cm
‑
fr.1和cm
‑
fr.2,分别上样;静置5h;
112.(8)使用蒸馏水洗脱,流速为0.5ml/min,每管收集1ml,每种洗脱剂收集60管;苯酚硫酸法跟踪检测,使用紫外分光光度计于490nm处测定吸光值;分别以管数、od 490nm作为横、纵坐标,绘制洗脱曲线;分别合并cm
‑
fr.1和cm
‑
fr.2洗脱过程中对应的洗脱液;减压浓缩;于真空条件下冷冻干燥48h,得香橼果实精制多糖cm
‑
1和cm
‑
2。
113.实施例6从香橼果实粗多糖(cm)制备精制多糖cm
‑
1和cm
‑2114.(1)deae sepharose fast flow预处理:称取deae sepharose fast flow 80g于23℃去离子水浸泡30h,间歇搅拌;再使用5%稀盐酸浸泡30min,去离子水反复冲洗至酸度计测得溶液ph=7;使用5%naoh溶液浸泡30min,去离子水反复冲洗至酸度计测得溶液ph=8~9;超声除气泡;
115.(2)将(1)预处理的deae sepharose fast flow装入层析柱;恒流泵平衡进出口流速;
116.(3)使用去离子水充分溶解实施例3得到的香橼果实粗多糖(cm)并配置成浓度为13mg/ml的多糖溶液;多糖溶液经0.22μm微孔滤膜过滤;使用凝胶滴管均匀上样;静置8h;
117.(4)依次使用去离子水,0.05mol/l的nacl溶液洗脱,流速为1.2ml/min,每管收集4ml,每种洗脱剂收集60管;苯酚硫酸法跟踪检测,使用紫外分光光度计于490nm处测定吸光值;分别以管数、od 490nm作为横、纵坐标,绘制洗脱曲线;分别合并不同洗脱剂对应的洗脱液;50℃减压浓缩,将浓缩糖液置于透析袋(截留量=8kda)于0℃蒸馏水中透析72h;于真空条件下冷冻干燥36h得到cm
‑
fr.1和cm
‑
fr.2;
118.(5)交联葡聚糖凝胶sephadex g
‑
100预处理:使用去离子水于80℃浸泡sephadex g
‑
1004h;20r/min磁力搅拌40min;置于20℃冷却;
119.(6)将经预处理的sephadex g
‑
100凝胶脱气;装入层析柱;恒流泵平衡进出口流速;
120.(7)使用去离子水充分溶解cm
‑
fr.1和cm
‑
fr.2,分别配置成5mg/ml的多糖溶液;多糖溶液经0.22μm微孔滤膜过滤;分别上样;静置8h;
121.(8)使用蒸馏水洗脱,流速为0.3ml/min,每管收集1.5ml,每种洗脱剂收集50管;苯酚硫酸法跟踪检测,使用紫外分光光度计于490nm处测定吸光值;分别以管数、od 490nm作为横、纵坐标,绘制洗脱曲线;分别合并cm
‑
fr.1和cm
‑
fr.2洗脱过程中对应的洗脱液;减压浓缩;于真空条件下冷冻干燥36h,得香橼果实精制多糖cm
‑
1和cm
‑
2。
122.实施例7香橼果实精制多糖cm
‑
1和cm
‑
2的分子量分布及均质性分析
123.采用高效凝胶渗透色谱法(hpgpc)分别对实施例4~6制备的cm
‑
1和cm
‑
2进行分子量测定并分析其均质性。色谱条件为:tsk g
‑
5000
pwxy column(7.8
×
300mm)和sk g
‑
3000
pwxy column(7.8
×
300mm)串联,流动相为0.02mol/l kh2po4缓冲溶液(ph=6),流速为0.6ml/min,2414示差检测器,柱温35℃。图3是实施例4制备得到的cm
‑
1和cm
‑
2的高效凝胶渗透色谱图(gpc);其中a为cm
‑
1的hpgpc图,b为cm
‑
2的hpgpc图。
124.结果分析:实施例4制备得到的cm
‑
1(图3中a)和cm
‑
2(图3中b)的gpc图均为单一对称的尖峰,表明分子量相对均一;cm
‑
1重均平均分子量(mw)为21520da,多分散性指数(pdi)为1.227493;cm
‑
2重均平均分子量(mw)为22303da,多分散性指数(pdi)为1.219320。实施例5和6结果同实施例4。
125.实施例8香橼果实精制多糖cm
‑
1和cm
‑
2的傅里叶红外光谱扫描(ft
‑
ir)
126.分别取实施例4~6中制备得到的干燥香橼果实精制多糖cm
‑
1和cm
‑
2各2mg,与kbr混合研磨成均匀并压片,于4000~400cm
‑1范围内进行红外扫描。图4是实施例4制备所得香橼果实精制多糖cm
‑
1和cm
‑
2的ft
‑
ir。
127.多糖红外光谱分析结果:
128.o
‑
h的伸缩振动引起3341cm
‑1(cm
‑
1)和3420cm
‑1(cm
‑
2)附近出现宽峰。c
‑
h的伸缩振动引起cm
‑
1和cm
‑
2分别在2933cm
‑1和2927cm
‑1的吸收峰。cm
‑
1和cm
‑
2在1750cm
‑1附近均没有信号峰,表明两种纯化多糖不含有酯化羧基。cm
‑
1和cm
‑
2在1646cm
‑1和1651cm
‑1处的吸收峰可能是由于o
‑
h形变振动引起,这可能归因于cm
‑
1和cm
‑
2极易吸潮而导致供试样品含有水分;样品和kbr在ft
‑
ir试验前要尽量保持干燥。1660cm
‑1,1538cm
‑1,1290cm
‑1表示酰胺i,ii,iii的特征吸收,而两种纯化组分的红外图谱在上述3处均没有出峰,表明样品中不含有蛋白质。1422cm
‑1(cm
‑
1)和1394cm
‑1(cm
‑
2)附近的信号峰归属于c
‑
h弯曲振动。cm
‑
2在1260cm
‑1处信号是s
‑
o非对称拉伸振动的特征吸收。cm
‑
1和cm
‑
2在1200
‑
1000cm
‑1的吸收带是由于c
‑
o
‑
c和c
‑
o
‑
h伸缩振动引起。一般认为,896cm
‑1(cm
‑
2)的特征吸收与β
‑
构型有关;858cm
‑1(cm
‑
1)和856cm
‑1(cm
‑
2)的特征信号与呋喃糖有关。α
‑
构型多糖引起807cm
‑1和810cm
‑1的特征吸收。1087cm
‑1,620cm
‑1,574cm
‑1(cm
‑
1)以及603cm
‑1(cm
‑
2)表示存在吡喃糖碳骨架。ft
‑
ir结果表明,cm
‑
1和cm
‑
2具有多糖官能团的特征吸收。实施例5和实施例6的检测结果同实施例4。
129.实施例9香橼果实精制多糖cm
‑
1和cm
‑
2的甲基化分析
130.取实施例4中制备得到的干燥香橼果实精制多糖cm
‑
1和cm
‑
2各15mg,分别加入6mldmso至完全溶解后,加入200mg naoh并超声3h至溶液呈淡黄色;冰水浴下缓慢滴加碘甲烷4ml,室温避光磁力搅拌48h。4℃下蒸馏水透析48h(截留量为3000da),使用二氯甲烷萃取并收集二氯甲烷相,蒸馏水洗涤3次后,55℃加压蒸干,加入甲醇反复洗涤并蒸干。取干燥样品进行ft
‑
ir检测,重复实验至3300cm
‑1~3450cm
‑1‑
oh吸收完全消失,甲基化结束。向cm
‑
1和cm
‑
2的甲基化干品中加入4ml 2mol/l三氟乙酸,酒精喷灯封口,于110℃水解8h至溶液呈现淡黄色,终止酸水解。分别向cm
‑
1和cm
‑
2的酸水解液中加入naoh调节ph至10~12,加入nabh
4 100mg避光反应12h,使用冰醋酸调节ph至中性;55℃减压蒸干,并使用甲醇反复洗涤后蒸干。向cm
‑
1和cm
‑
2还原之后的干品中按1:1(v∶v)加入吡啶和醋酸酐,于沸水浴中磁力搅拌1h,过0.22μm微孔滤膜,使用gc
‑
ms进行分析。
131.cm
‑
1和cm
‑
2甲基化分析的结果如表1所示。cm
‑
1具有8种不同的糖苷键,阿拉伯糖(46.35%)与木糖(46.64%)的含量共占总糖基的92.99%。cm
‑
2具有7种不同的糖苷键,阿拉伯糖(79.57%)与半乳糖(15.51%)的含量共占总糖基的95.08%,这与单糖组成得出的结果一致。cm
‑
1和cm
‑
2末端残基的含量分别高达40.34%和32.19%;且两者末端残基与分支点的个数大致相等(比值约为1)。根据公式1得出cm
‑
1和cm
‑
2的分支度(db)分别高达75.03%和51.31%。
132.db=(n
t
+n
b
)/(n
t
+n
b
+n
l
)
ꢀꢀꢀ
(公式1)
133.其中n
t
,n
b
,n
l
分别代表末端残基,支链残基和线状残基的摩尔比。
134.表1 cm
‑
1和cm
‑
2甲基化分析结果
[0135][0136][0137]
实施例10香橼果实精制多糖cm
‑
1的核磁共振分析(1d,2d nmr)和化学结构
[0138]
40mg cm
‑
1分别溶解于0.8ml d2o,冻溶3次,0.8ml d2o溶解,转入核磁管。600mhznmr光谱仪于298k记录1h nmr,
13
c nmr,hsqc,1h
‑1h cosy,hmbc光谱。
[0139]
cm
‑
1的nmr分析结果如图5所示。图5是实施例4得到的cm
‑
1的核磁共振图谱(nmr)和化学结构;a是1h nmr,b是
13
c nmr,c是hsqc,d是cosy,e是hmbc,f是化学结构。根据各表征及分析确定表1中糖苷键的α/β构型,并确定异头氢的位移,通过hsqc对异头碳的信号进行归属;再利用1h
‑1h cosy对其他h进行信号归属,同样利用hsqc对其余碳信号进行完整归属并于相应位置进行标记。根据hmbc确定不同糖残基的连接序列,还原糖链结构如图5中f所示。
[0140]
cm
‑
1是一种新型的阿拉伯木聚糖;以1,4
‑
β
‑
d
‑
xylp为主链,在o
‑
3位被α
‑
l
‑
araf单取代,其中包括α
‑
(1,3)或α
‑
(1,5)连接的阿拉伯糖短侧链以及α
‑
t
‑
araf。先前报道中,阿拉伯木聚糖一般来源于谷类如小麦,大麦等,于其他药食同源的植物多糖中鲜有报道。且已报道的阿拉伯木聚糖一般是于木糖的o
‑
2和o
‑
3位置均被阿拉伯糖基取代;鲜有如cm
‑
1仅于o
‑
3位置取代的阿拉伯木聚糖。
[0141]
实施例11香橼果实精制多糖cm
‑
2的核磁共振分析(1d,2d nmr)
[0142]
40mg cm
‑
2分别溶解于0.8ml d2o,冻溶3次,0.8ml d2o溶解,转入核磁管。600mhznmr光谱仪于298k记录1h nmr,
13
c nmr,hsqc,1h
‑1h cosy,hmbc光谱。
[0143]
cm
‑
2的nmr分析结果如图6所示。图6是实施例4得到的cm
‑
2的核磁共振图谱(nmr)和化学结构;a为1h nmr,b为
13
c nmr,c为hsqc,d为cosy,e为hmbc,f为化学结构。根据各表征及分析,得出表1中糖苷键的α/β构型,并确定异头氢的位移,通过hsqc对异头碳的信号进行归属;再利用1h
‑1h cosy对其他h进行信号归属,同样利用hsqc对其余碳信号进行完整归属并于相应位置进行标记。根据hmbc确定不同糖残基的连接序列,还原糖链结构如图6中f所示。
[0144]
cm
‑
2是一种新型的半乳阿拉伯聚糖,以(1
→
5)
‑
α
‑
l
‑
araf为主链,在o
‑
2或/和o
‑
3位置主要被β
‑
d
‑
galp取代。先前报道中,富含半乳糖和阿拉伯糖的多糖通常是第一类或第二类阿拉伯半乳聚糖,鲜有如cm
‑
2因1.2,3,5
‑
α
‑
l
‑
araf含量高而分支度高的半乳阿拉伯聚糖。
[0145]
实施例12香橼果实精制多糖cm
‑
1和cm
‑
2的扫描电镜图(sem)
[0146]
分别取实施例4~6中制备得到的干燥香橼果实精制多糖cm
‑
1和cm
‑
2各2mg于固定的金属台减压镀金,加速电压5.0kv,高真空环境,使用扫描电子显微镜(sem)观察两者表面样貌。图像放大倍率设置为200
×
,500
×
和1000
×
。图7是实施例4制备所得香橼果实精制多糖cm
‑
1和cm
‑
2的sem图;其中a为cm
‑
1的sem图,b为cm
‑
2的sem图。
[0147]
多糖电镜扫描结果分析:
[0148]
cm
‑
1和cm
‑
2皆呈聚集状态。cm
‑
1表面多孔,cm
‑
2呈片状且少有缝隙。相比而言,cm
‑
1形貌更松散,且卵状颗粒的密集分布使其表面更加粗糙。实施例5和实施例6的检测结果同实施例4。
[0149]
实施例13香橼果实精制多糖cm
‑
1和cm
‑
2的热重
‑
差示扫描热量同步热分析(tg
‑
dsc)
[0150]
分别取实施例4~6中制备得到的干燥香橼果实精制多糖cm
‑
1和cm
‑
2各5mg置于托盘中,于n2氛围中以10℃/min的升温速率从30℃升至800℃。图8是实施例4得到的cm
‑
1和cm
‑
2的热稳定性分析图(tg
‑
dsc)。
[0151]
多糖热稳定性结果分析:
[0152]
在dsc曲线中,向下的峰表示δh<0(放热反应)。从室温到800℃,cm
‑
1的质量变化主要经历了2个阶段:第一阶段是失水阶段,主要失去物理吸附的水分(冻干残留水分),质量损失3.65%,最大失重速率对应的温度为84.7℃;第二阶段是热解阶段(包括热解聚,热分解和热降解),质量减少61.53%,最大热分解速率出现在270.6℃。最后,cm
‑
1残余的质量为34.82%。然而,cm
‑
2的质量变化主要经历3个阶段:失水阶段,主要热解阶段和残碳分解(碳化)阶段。第一阶段(失水)质量损失为0.36%,最大失重速率对应的温度为74.63℃;第二阶段(主要热解阶段)质量减少50.15%%,最大热分解速率出现在306.2℃;第三阶段质量损失为4.18%,可归因于在较低温度下产生的碳残留物进一步分解,最大热分解速率出现在744℃。最终,cm
‑
2残余质量为44.68%。相比而言,第一阶段cm
‑
1质量损失较大,说明其持水力较强。cm
‑
2的最大热解温度较高,且最终残余质量高于cm
‑
1,这表明cm
‑
2的热稳定性高于cm
‑
1,且两种多糖组分的降解行为存在较大差异。
[0153]
实施例14香橼果实精制多糖cm
‑
1和cm
‑
2对α
‑
淀粉酶抑制作用
[0154]
表2 cm
‑
1和cm
‑
2对α
‑
淀粉酶抑制作用的反应体系
[0155][0156]
溶液配制:(1)pbs(ph=7.4):酸度计调节0.1mol/l nah2po4、0.1mol/l na2hpo4至混合液ph=7.4。
[0157]
(2)5u/ml α
‑
淀粉酶溶液:10mg α
‑
淀粉酶(5u/mg)溶解于10ml pbs(ph=7.4),
‑
20℃保存7天。临用前,37℃活化35min。
[0158]
(3)1%淀粉溶液:100g沸水溶解1g淀粉,搅拌,冷却封口。
[0159]
(4)10mg/ml二硝基水杨酸溶液:100ml超纯水溶解3,5
‑
二硝基水杨酸1g,避光4℃保存。
[0160]
(5)样品及阳性对照:超纯水制备cm
‑
1和cm
‑
2溶液和阿卡波糖(阳性对照)溶液(3.2,1.6,0.8,0.4,0.2mg/ml)各3ml,备用。
[0161]
检测方法:试验设置样品组,阳性对照组,空白组,背景组。
[0162]
在容器中按照表2加入各物质,25℃恒温孵育10min,加入50μl质量分数为1%淀粉溶液,25℃恒温孵育10min;加入100μl的3,5
‑
二硝基水杨酸溶液置于沸水浴10min,冷却。最后,使用酶标仪于405nm检测吸光值。根据公式2计算抑制率:
[0163]
样品/阳性对照对α
‑
淀粉酶抑制率(%)=[1
‑
(a
样品组
‑
a
背景组
)/a
空白组
]
×
100%
ꢀꢀꢀ
(公式2)
[0164]
cm
‑
1和cm
‑
2对α
‑
淀粉酶抑制作用结果分析:
[0165]
结果如图9所示,图9是实施例4制备所得香橼果实精制多糖cm
‑
1和cm
‑
2对α
‑
淀粉酶抑制率的曲线图。cm
‑
1,cm
‑
2及阿卡波糖均以浓度依赖性抑制α
‑
淀粉酶活性,并于3.2mg/ml抑制率达到最高,分别为47.93%
±
3.25%,62.00%
±
4.50%,和99.06%
±
5.77%。结果显示实施例4制备所得香橼果实精制多糖cm
‑
1和cm
‑
2对α
‑
淀粉酶具有抑制作用。
[0166]
实施例15香橼果实精制多糖cm
‑
1和cm
‑
2对α
‑
葡萄糖苷酶抑制作用
[0167]
表3 cm
‑
1和cm
‑
2对α
‑
葡萄糖苷酶抑制作用的反应体系
[0168][0169]
其中,溶液配制:(1)pbs(ph=6.8):酸度计调节0.1mol/l nah2po4、0.1mol/l na2hpo4至混合液ph=6.8。
[0170]
(2)1u/ml α
‑
葡萄糖苷酶溶液:100mgα
‑
葡萄糖苷酶(100u/g)溶解于10ml pbs(ph=6.8),
‑
20℃封口保存。临用前,37℃活化35min。
[0171]
(3)10mmol/l对硝基苯酚
‑
α
‑
d
‑
葡萄糖苷(pnpg)溶液:25ml pbs(ph=6.8)溶解
pnpg 75.312mg,4℃避光保存。
[0172]
(4)1mol/lna2co3溶液:50ml超纯水溶解na2co
3 5.29g,密封保存。
[0173]
(5)样品及阳性对照:超纯水制备cm
‑
1和cm
‑
2溶液和阿卡波糖(阳性对照)溶液(3.2,1.6,0.8,0.4,0.2mg/ml)各3ml,备用。
[0174]
检测方法:试验设置样品组,阳性对照组,空白组,背景组。
[0175]
在反应装置中按照表3加入各物质,37℃恒温孵育30min,再加入50μl pnpg溶液,37℃恒温孵育10min,最后再加入100μl na2co3溶液,于405nm测定其吸光值。根据公式3计算抑制率:
[0176]
样品/阳性对照对α
‑
葡萄糖苷酶抑制率(%)=[1
‑
(a
样品组
‑
a
背景组
)/a
空白组
]
×
100%
ꢀꢀꢀ
(公式3)。
[0177]
cm
‑
1和cm
‑
2对α
‑
葡萄糖苷酶抑制作用结果分析:图10是实施例4制备所得香橼果实精制多糖cm
‑
1和cm
‑
2对α
‑
葡萄糖苷酶抑制率的曲线图。
[0178]
cm
‑
1,cm
‑
2及阿卡波糖均以浓度依赖性抑制α
‑
葡萄糖苷酶活性,且于3.2mg/ml抑制率达到最高,分别为36.36%
±
2.05%,56.11%
±
2.89%和99.49%
±
5.45%。结果显示实施例4制备所得香橼果实精制多糖cm
‑
1和cm
‑
2对α
‑
葡萄糖苷酶具有抑制作用。
[0179]
实施例16香橼果实精制多糖cm
‑
1和cm
‑
2对胰岛素抵抗
‑
肝癌细胞(hepg2
‑
ir)葡萄糖消耗量的影响
[0180]
使用mtt法测定cm
‑
1和cm
‑
2对正常hepg2细胞存活率的影响。将hepg2细胞以每孔100μl密度为8
×
104个/ml接种在96孔板上。孵育24h后,用不同浓度已灭菌的cm
‑
1,cm
‑
2溶液(0、50、100、200、400μg/ml)作用24h。然后,弃去培养基,每孔加入200μlmtt溶液(0.5mg/ml,溶解于dmem)。4h后,弃去上清液,每孔加入150μl dmso溶解已生成的甲瓒。最后,使用酶标仪在490nm处测定吸光值。抑制率按公式4计算:
[0181]
细胞增殖抑制率(%)=[1
‑
a2/a1]
×
100%
ꢀꢀꢀ
(公式4)
[0182]
a1是没添加样品的吸光值。a2是试验样品组的吸光值。
[0183]
采用“高糖+胰岛素”诱导hepg2
‑
ir。具体步骤包括:细胞铺板,诱导,检测上清液葡萄糖含量,mtt法测定诱导后细胞存活率。按照表4向96孔板(以100μl/孔接种)中加入各物质(其中,hepg2细胞悬液是由hepg2细胞均匀分散于dmem完全培养基而制得),孵育24h,弃去上清液,空白组和正常组中加入100μl dmem完全培养基,模型组中加入100μl胰岛素(1
×
10
‑7mol/l,,溶解至dmem完全培养基);孵育36h,弃上清液,三组分别加入100μl dmem基础培养基,孵育12h,上清液检测葡萄糖含量,细胞使用mtt法测定存活率。按照葡萄糖检测试剂盒(god
‑
pod)说明书规范操作,使用酶标仪于560nm测定吸光值。根据公式5,6测定上清葡萄糖消耗量。对比模型组与正常组上清葡萄糖含量,判断hepg2
‑
ir建模是否成功。样品组、阳性对照组是向hepg2
‑
ir细胞加入不同浓度cm
‑
1,cm
‑
2(400,200,100,50μg/ml)或2mmol/l二甲双胍溶液(溶于dmem基础培养基)。孵育24h,收集上清,利用公式5,6检测葡萄糖含量。
[0184]
表4 hepg2
‑
ir细胞建模
[0185]
[0186]
葡萄糖含量(mmol/l)=(a1/a2)
×
c0ꢀꢀꢀ
(公式5)
[0187]
葡萄糖消耗量=m0‑
m1ꢀꢀꢀ
(公式6)
[0188]
其中,a0,a1分别是于560nm测得的校准液和各组上清液的吸光值;c0是校准液浓度。m0是dmem(完全)葡萄糖含量,m1是各组细胞上清中葡萄糖含量。
[0189]
试验结果如图11所示,图11是实施例4得到的cm
‑
1和cm
‑
2对胰岛素抵抗
‑
肝癌细胞(hepg2
‑
ir)葡萄糖消耗量影响图以及对正常hepg2细胞和hepg2
‑
ir细胞存活率影响图;a是cm
‑
1和cm
‑
2对正常hepg2细胞存活率影响的柱形图;b是cm
‑
1和cm
‑
2对hepg2
‑
ir细胞存活率影响的柱型图;c是cm
‑
1和cm
‑
2对hepg2
‑
ir细胞葡萄糖消耗量影响的柱形图。
[0190]
根据图11中a,浓度范围为50~400μg/ml的cm
‑
1和cm
‑
2作用于正常hepg2细胞24h后,细胞存活率均在95%~100%之间,表明cm
‑
1和cm
‑
2对正常hepg2细胞无毒性,且不会过度促进细胞增殖,cm
‑
1和cm
‑
2(50~400μg/ml)可用于进行后续降糖试验的研究。
[0191]
根据图11中c,模型组(mod)hepg2细胞葡萄糖消耗量为(0.64
±
0.031)mmol/l,为正常对照组(ngt)的40.86%;且根据图11中b可知,hepg2
‑
ir细胞的存活率在96%~100%,表明细胞建模成功;以2mmol/l二甲双胍(dmbg)为阳性对照,cm
‑
1,cm
‑
2以剂量依赖性增加葡萄糖的消耗量,于400μg/ml浓度时,葡萄糖消耗量分别为0.96
±
0.082,1.33
±
0.050;相比于模型组分别增加0.31,0.68mmol/l;葡萄糖消耗量分别占正常对照组的60.36%,84.03%。根据图11中a和b可排除cm
‑
1、cm
‑
2和诱导方式对细胞存活率的影响,也进一步说明50~400μg/ml香橼果实精制多糖cm
‑
1,cm
‑
2通过修复hepg2
‑
ir细胞来发挥降糖功效。
[0192]
实施例17香橼果实精制多糖cm
‑
1和cm
‑
2对uvb损伤的hacat细胞的修复作用
[0193]
首先,使用mtt法测定浓度范围为50~400μg/ml的cm
‑
1和cm
‑
2对正常hacat细胞存活率的影响。
[0194]
然后,建立uvb致损伤的hacat模型,使用cm
‑
1和cm
‑
2修复。正常组、模型组和样品组中,将hacat细胞以每孔100μl密度为2
×
105个/ml接种在96孔板上。孵育24h后,弃去上清,pbs洗板3次,加入100μlpbs,正常组以锡箔纸覆盖,模型组和样品组无锡箔纸覆盖;60mj/cm
2 uvb处理;样品组中加入50~400μg/ml cm
‑
1或cm
‑
2溶液,正常组和模型组中无多糖,孵育24h,使用mtt法测定细胞存活率。
[0195]
测试结果如图12所示,图12是实施例4得到的cm
‑
1和cm
‑
2对uvb损伤的hacat细胞修复的影响以及对正常的hacat细胞存活率影响;a是cm
‑
1和cm
‑
2对正常的hacat细胞存活率影响的柱形图;b是cm
‑
1和cm
‑
2对uvb损伤的hacat细胞修复影响的柱形图。根据图12中a,50~400μg/ml的cm
‑
1和cm
‑
2作用正常hacat细胞24h,细胞存活率均在96%~100%之间;表明cm
‑
1和cm
‑
2对正常hacat细胞无毒性,且不会过度促进细胞增殖,cm
‑
1和cm
‑
2(50~400μg/ml)可用于进行后续抗皮肤光老化试验的研究。
[0196]
经过uvb能量的筛选,60mj/cm2对正常hacat细胞的致死率在50%左右,为最佳能量照射值。根据图12中a,50~400μg/ml浓度范围内的cm
‑
1和cm
‑
2不会引起hacat细胞过度增殖,也进一步表明香橼果实精制多糖cm
‑
1和cm
‑
2对60mj/cm2uvb照射致损伤的hacat细胞具有修复效果。
[0197]
实施例18香橼果实精制多糖cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7的激活作用
[0198]
使用mtt法测定浓度范围为50~400μg/ml cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7存活率的影响,以lps(1μg/ml)为阳性对照。同时,于倒置显微镜下观察并拍照空白对照、
400μg/ml cm
‑
1和cm
‑
2、1μg/ml lps作用raw264.7细胞24h后细胞形态的变化。
[0199]
测试结果如图13所示;图13是实施例4得到的cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7的激活作用;a是cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7存活率影响的柱形图;b是cm
‑
1和cm
‑
2激活小鼠巨噬细胞raw264.7倒置显微镜拍照图。
[0200]
根据图13中a,50~400μg/ml cm
‑
1和cm
‑
2以及阳性对照lps(1μg/ml)作用raw264.7细胞24h后,细胞存活率在97%~100%之间,无细胞毒性,可用于后续免疫增强试验的研究。根据图13中b,空白对照组细胞呈圆形,体积较小且聚团生长;而经400μg/ml cm
‑
1和cm
‑
2处理后的raw264.7细胞伸出伪足变为长梭形,细胞体积增大,生长相对分散,且贴壁生长;这些变化与阳性对照组(lps)表现一致。由此表明,cm
‑
1和cm
‑
2和lps同样具有激活巨噬细胞的效果。经显色基质鲎试剂盒(lal)检测表明cm
‑
1和cm
‑
2未经内毒素污染,表明免疫细胞被激活是由于样品本身的刺激作用所致。
[0201]
实施例19香橼果实精制多糖cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7吞噬中性红的影响
[0202]
将密度为2
×
105个/ml的raw246.7细胞以每孔100μl接种在96孔板上,孵育24h后,加入cm
‑
1,cm
‑
2(0、50、100、200、400μg/ml)和阳性对照lps(1μg/ml)作用24h。收集上清备用,向每孔加入150μl 0.1%中性红溶液(溶解在pbs中)。1h后,用pbs洗板3次,向每孔加入150μl细胞裂解液(乙酸∶乙醇=1∶1,v/v)。避光反应2h,于550nm处测定吸光值。吞噬指数(pi)由公式7计算得出:
[0203]
吞噬指数(pi)%=(a2/a1)
×
100%
ꢀꢀꢀ
(公式7)
[0204]
测试结果如图14所示,图14是实施例4得到的cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7吞噬中性红影响的柱形图。其中,p<0.05具有统计学意义,每列中不同小写字母表示具有显著性差异(p<0.05)。cm
‑
1和cm
‑
2均能显著促进小鼠巨噬细胞raw264.7吞噬中性红的能力,400μg/ml的cm
‑
1促进效果与阳性对照相当。
[0205]
实施例20香橼果实精制多糖cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7释放tnf
‑
α,il
‑
6和no的影响
[0206]
将密度为2
×
105个/ml的raw 246.7细胞以每孔100μl接种在96孔板上,孵育24h后,加入cm
‑
1,cm
‑
2(0、50、100、200、400μg/ml)和阳性对照lps(1μg/ml)作用24h。收集上清备用,按照tnf
‑
α、il
‑
6elisa,griess试剂盒说明书规范操作。
[0207]
测试结果如图15所示,图15是实施例4得到的cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7释放tnf
‑
α,il
‑
6和no影响的柱形图;a是cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7释放tnf
‑
α影响的柱形图;b是cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7释放il
‑
6影响的柱形图;c是cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7释放no影响的柱形图。其中,p<0.05具有统计学意义,每列中不同小写字母表示具有显著性差异(p<0.05)。与空白对照相比,cm
‑
1和cm
‑
2在100~400μg/ml浓度范围内均能显著促进raw264.7细胞分泌tnf
‑
α,il
‑
6,no(p<0.05);其中,400μg/ml cm
‑
1促进raw264.7细胞分泌tnf
‑
α,il
‑
6与阳性对照效果相当。
[0208]
实施例21香橼果实精制多糖cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7的tnf
‑
α,il
‑
6和inos mrna表达的影响
[0209]
rt
‑
qpcr法检测cm
‑
1和cm
‑
2对raw264.7细胞tnf
‑
α,il
‑
6,inos mrna表达量的影响,主要步骤包括:总rna提取,测定rna浓度和纯度,rna逆转录,rt
‑
qpcr扩增。
[0210]2×
106个/ml raw246.7细胞以500μl/孔接种于24孔板。孵育20h,弃上清,分别加入500μl不同终浓度的cm
‑
1,cm
‑
2(0、50、100、200、400μg/ml)或lps(1μg/ml)。12h后,裂解细胞,trizol reagent提取总rna,记录od 260nm和od 280nm检测浓度和纯度。然后,将rna反转录成cdna。最后,采用rt
‑
qpcr定量分析相关基因mrna,所用引物列于表5中。gapdh作内参,2
‑
δδct
方法进行分析。
[0211]
表5 rt
‑
qpcr中所用引物序列
[0212][0213]
测试结果如图16所示,图16是实施例4得到的cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7的tnf
‑
α,il
‑
6和inos mrna表达影响的柱形图;a是cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7的tnf
‑
α mrna表达量影响的柱形图;b是cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7的il
‑
6 mrna表达量影响的柱形图;c是cm
‑
1和cm
‑
2对小鼠巨噬细胞raw264.7的inos mrna表达量影响的柱形图。其中,p<0.05具有统计学意义,每列中不同小写字母表示具有显著性差异(p<0.05)。与空白对照相比,浓度范围为100~400μg/ml的cm
‑
1和cm
‑
2均可显著促进tnf
‑
α,il
‑
6,inos mrna(p<0.05)。由此推测,cm
‑
1和cm
‑
2可能通过上调raw264.7细胞tnf
‑
α,il
‑
6,inos mrna的表达来促进tnf
‑
α,il
‑
6,no的释放进而发挥免疫增强效果。综合实例14~17可得,cm
‑
1和cm
‑
2具有免疫增强功效。
[0214]
cm
‑
1是一种新型的阿拉伯木聚糖;以1,4
‑
β
‑
d
‑
xylp为主链,在o
‑
3位被α
‑
l
‑
araf单取代,其中包括α
‑
(1,3)或α
‑
(1,5)连接的阿拉伯糖短侧链以及α
‑
t
‑
araf。据报道谷物来源的阿拉伯木聚糖,在木糖的o
‑
2和o
‑
3位或者仅o
‑
2位置被阿拉伯糖基取代,呈现出较好的免疫调节性。但cm
‑
1是仅于木糖的o
‑
3位置被阿拉伯糖基取代,且侧链1.2,3,5
‑
α
‑
l
‑
araf的存在使cm
‑
1具有高分支度和更复杂的结构;上述结构特征可能是cm
‑
1呈现较好的免疫刺激性的原因。
[0215]
cm
‑
2是一种新型的半乳阿拉伯聚糖,以(1
→
5)
‑
α
‑
l
‑
araf为主链,在o
‑
2或/和o
‑
3位置主要被β
‑
d
‑
galp取代。据报道二型阿拉伯半乳聚糖具有良好的免疫活性,而半乳阿拉伯聚糖的免疫活性未有涉及。本发明首次证明半乳阿拉伯聚糖如cm
‑
2具有免疫增强活性,结合cm
‑
1的结构特征,免疫增强活性可能与适度含量的阿拉伯糖基、高分支度、骨架结构、侧链结构(取代位置,取代度,取代基团)等有关。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1