一种螺旋超分子材料及其制备方法与应用
本发明属于超分子化学技术领域,涉及一种多螺旋超分子结构材料的合成,可应用于对杯[n]芳烃(n=4-8)进行主客体识别。
背景技术:
超分子化学这一概念早在上个世纪70年代被提出,现如今已经发展成为了化学领域中的一个重要分支。非共价相互作用是研究超分子化学的基础,包含氢键、离子键、π-π堆积、配位键、亲疏水等。其中,基于金属-配体配位驱动的分子自组装由于合适的键能和导向性,已经成为构筑精准离散超分子组装体最有效的策略之一。对于多吡啶配体以及吡啶的各种衍生物来说,由于其中的氮原子含有固定的孤对电子,所以吡啶基配体能够与多种金属盐产生配位作用,吡啶类超分子结构大大丰富了超分子体系的内容,是配位化学中经典的体系之一。其中一种被经常使用的吡啶类构建基元就是三联吡啶(tpy),三联吡啶衍生物由于它们对不同的裸过渡金属离子有较强的结合能力,在超分子自组装领域引起相当大的关注。目前,通过三联吡啶基配体与金属离子的配位已经成功构建了许多二维的结构,并且这些结构的复杂性和新颖性也在不断提高。但是由于三联吡啶和金属离子的配位角度一般固定在180°,所以通过设计基于三联吡啶的配体使其与金属配位成精准可控的三维超分子结构仍是一个难题,这限制了三联吡啶配位在三维超分子结构中的应用。在本设计中,设计了一种多齿三联吡啶配体,为了减少三联吡啶八面体配位的空间位阻,将三联吡啶单元设计到了最终结构的中心。通过这些配体和过渡金属的组装成功合成了一系列三维螺旋超分子结构。
在众多超分子体系中,主客体识别体系在过去几十年中受到了广泛的关注,主客体识别的应用包括对特定客体分子的靶向识别、混合物的纯化以及产生新的不对称催化途径等。其中,三维的金属有机超分子体系由于结构的稳定性好,具有特殊的内部腔体和易修饰的官能团配体,所以,设计合适的组装基元,精准调控三维分子笼的腔体,使其可以特异性识别并封装指定的客体是三维金属有机超分子体系的研究热点之一。在超分子客体分子的封装研究中,绝大多数的研究把客体集中在规则结构上,例如聚芳烃和富勒烯,很少有报道把具有复杂构象的杯芳烃作为客体的研究。所以,利用三维金属有机超分子对杯芳烃进行主客体识别的研究有着很大的价值。
三维金属有机超分子的主客体识别主要原理:首先三维金属有机超分子提供合适大小的空腔与适当的空间限制来容纳及约束客体分子,其次超分子主体和客体之间通过一种或几种非共价作用力形成主客体结构,所涉及的作用力包括π-π作用力、静电作用力和疏水作用力等。三维金属有机超分子的空腔和客体之间良好的尺寸和形状匹配十分重要,主体空腔狭小会使客体在三维超分子主体内运动受到限制,而主体太大的空腔将减弱主客体间的范德华作用力及静电吸引力。所以,针对不同的大小、形状的客体要通过具有合适空腔大小的三维超分子与其发生主客体作用。
但已报导的主客体系统中,对于三维金属有机超分子主体的空腔大小和整体空间约束上的调节还存在不足,这导致了像杯芳烃这样具有复杂构象的客体无法被合适的主体识别,使得主客体系统不完善。
技术实现要素:
本发明要解决的技术问题是,克服现有主客体系统存在的不足,提供一种具有不同空间限制、不同空腔大小的三维多螺旋超分子材料,以及该多螺旋超分子材料的合成方法和作为主体特异性识别杯[n]芳烃(n=4-8)客体方面的应用。
上述的技术问题通过以下的技术方案实现:
一种螺旋超分子材料,是由多齿三联吡啶(tpy)配体分子和过渡金属m构成的螺旋结构配合物,具有如下的分子结构:
在上述结构中,m代表过渡金属,
作为优选,所述过渡金属m可以为zn、cd、fe、ru、hg、cu、ag、au、ni、pd、pt、co、rh、ir、os、mn、tc、re中的一种。
作为优选,
更优选的,所述多齿三联吡啶配体分子的结构中的取代基r为氢、烷氧基或醚氧基;r1、r2为氢或烷基。
一种螺旋超分子材料的制备方法,有以下步骤:
(1)化合物z的合成
多齿三联吡啶(tpy)配体分子的合成需要使用中间体化合物z,化合物z的合成路线为:
将化合物x、化合物y、pd(pph3)2cl2和碳酸钠以摩尔比1:1:0.05:6的比例混合在一起,在氮气氛围下以4:2:1的体积比加入甲苯、水和叔丁醇,将混合物在85℃下搅拌12小时,反应结束后用ch2cl2萃取,通过柱层析纯化粗产物得到化合物z;其中化合物x、化合物y的结构如下:
化合物x的合成方法参照文献yi-tsuchan,daltontrans.2015,44(11),5139-5145;
(2)多齿三联吡啶配体的合成
将化合物z、配体中心核化合物、pd(pph3)2cl2和碳酸钠混合在一起,在氮气氛围下以4:2:1的体积比加入甲苯、水和叔丁醇,将混合物在85℃下搅拌3-6天,反应结束后用chcl3萃取,通过柱层析纯化粗产物得到多齿三联吡啶配体;其中,当配体中心核化合物选自以下a、b、c之一时,化合物z、配体中心核化合物、pd(pph3)2cl2和碳酸钠的反应摩尔比为3:1:0.15:6,且最终得到的配体结构分别如a、b、c;当配体中心核化合物选自以下d、e、f、g之一时,化合物z、配体中心核化合物、pd(pph3)2cl2和碳酸钠的反应摩尔比为4:1:0.2:6,且最终得到的配体的结构分别如d、e、f、g;当配体中心核化合物选自以下h、i之一时,化合物z、配体中心核化合物、pd(pph3)2cl2和碳酸钠的反应摩尔比为5:1:0.25:6,且最终得到的配体的结构分别如h、i;当配体中心核化合物选自以下j、k之一时,化合物z、配体中心核化合物、pd(pph3)2cl2和碳酸钠的反应摩尔比为6:1:0.3:6,且最终得到的配体的结构分别如j、k;
(3)多螺旋超分子材料的合成
将步骤(2)中得到的多齿三联吡啶配体与过渡金属m盐溶于氯仿或极性溶剂中,在50-125℃的温度下,反应8-24h,反应结束后加不良溶剂析出,经离心得到多螺旋超分子;所述极性溶剂为ch3cn、dmso、ch3oh、dmf等;不良溶剂根据多螺旋超分子性质确定,可选自乙醚、水;当使用不同的多齿三联吡啶配体时,配体与过渡金属m的摩尔比分别为:三齿三联吡啶配体与过渡金属m的反应摩尔比为2:3,四齿三联吡啶配体与过渡金属m的反应摩尔比为2:4,五齿三联吡啶配体与过渡金属m的反应摩尔比为2:5,六齿三联吡啶配j体与过渡金属m的反应摩尔比为2:6。
一种螺旋超分子材料的用途,其特征在于,利用所述的螺旋超分子材料与杯[n]芳烃发生主客体识别,其中n为4~8的整数,操作方法为:将所述螺旋超分子材料与客体分子溶于极性溶剂中,对所得混合溶液进行超声处理,即可得到螺旋超分子包封杯[n]芳烃客体的主客体体系;所述极性溶剂为ch3cn、dmso、dmf等;所述杯[n]芳烃的结构如下,取代基r1、r2为氢或烷基;
有益效果:
1、本发明通过合成多齿三联吡啶配体,并将多齿三联吡啶配体与过渡金属组装,利用三联吡啶配位角度180°的特征,形成三维螺旋超分子结构。
2、本发明合成的螺旋超分子可识别并封装具有复杂分子构象的杯[n]芳烃(n=4-8),这在已报导的主客体研究中很少见。
3、本发明提供一种具有不同空腔大小、不同空间限制的多螺旋超分子,每个螺旋超分子可根据自身的结构特征去特异性识别客体分子,这使得主客体系统更加完善。
附图说明:
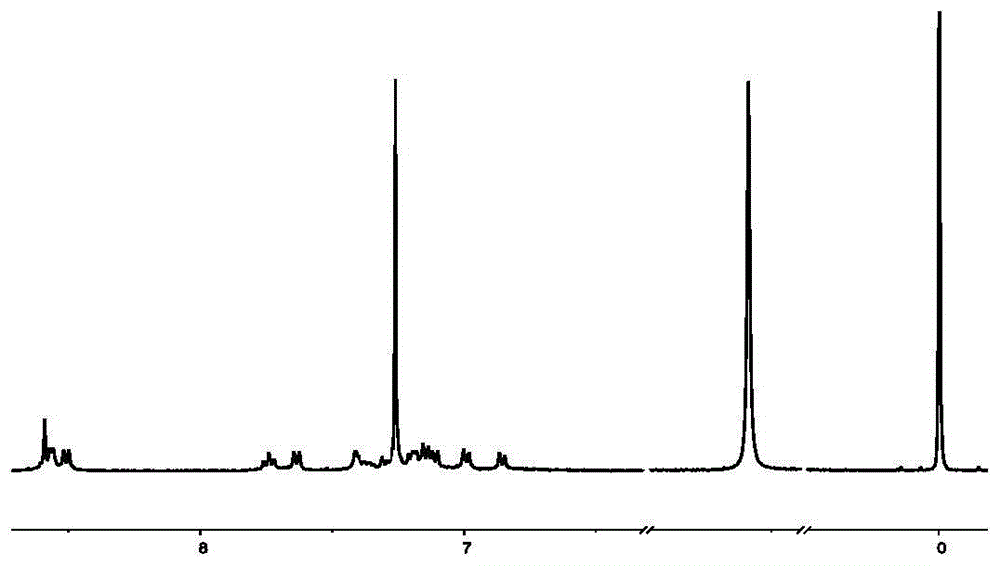
图1是实施例1制备的六齿三联吡啶配体la的1hnmr图。
图2是实施例1制备的六齿三联吡啶配体la的13cnmr图。
图3是实施例1制备的六齿三联吡啶配体la的2dcosynmr图。
图4是实施例1制备的六齿三联吡啶配体la的maldi-tof质谱图。
图5是实施例1制备的六螺旋超分子sa的1hnmr图。
图6是实施例1制备的六螺旋超分子sa的13cnmr图。
图7是实施例1制备的六螺旋超分子sa的2dcosynmr图。
图8是实施例1制备的六螺旋超分子sa的2ddosynmr图。
图9是实施例1制备的六螺旋超分子sa不同电荷的电喷雾质谱图(实验的同位素信号-下;模拟的同位素信号-上)。
图10是实施例1制备的六螺旋超分子sa的晶体结构图。
图11是实施例2中六螺旋超分子sa的1hnmr图(a)以及包封杯[6]芳烃后的主客体复合物sa2的1hnmr图(b)的对比。
图12是实施例2中主客体复合物sa2不同电荷的电喷雾质谱图(实验的同位素信号-下;模拟的同位素信号-上)。
图13是实施例3制备的三齿三联吡啶配体lb的1hnmr图。
图14是实施例3制备的三齿三联吡啶配体lb的maldi-tof质谱图。
图15是实施例3制备的三螺旋超分子sb的1hnmr图。
图16是实施例3制备的三螺旋超分子sb不同电荷的电喷雾质谱图(实验的同位素信号-下;模拟的同位素信号-上)。
图17是实施例4中三螺旋超分子sb的1hnmr图(a)以及包封杯[4]芳烃后的主客体复合物sb2的1hnmr图(b)的对比。
图18是实施例4中三螺旋超分子sb的1hnmr图(a)以及包封杯[6]芳烃后的主客体复合物sb3的1hnmr图(b)的对比。
具体实施方式
实施例1:六螺旋超分子sa的合成
六齿三联吡啶配体la的合成路线如下:
其中化合物1(xiaopengli,j.am.chem.soc.2017,139(24),8174-8185),化合物2(yi-tsuchan,daltontrans.2015,44(11),5139-5145),化合物4(mikioyasutake,tetrahedron2015,71(29),4714-4721),化合物5(pantelisn.trikalitis,j.am.chem.soc.2016,138(39),12767-12770)参照相关文献合成制备。
化合物3的合成:
将化合物2(2.0g,4.3mmol),1,4-苯二硼酸双(频哪醇)酯(4.3g,13.0mmol),pd(pph3)2cl2(151.4mg,0.22mmol)和碳酸钠(3.18g,30mmol)加入200mlschlenk烧瓶中。抽真空通氮气三次,并在氮气下加入甲苯(60ml),水(30ml)和叔丁醇(15ml)。将混合物在85℃下搅拌12小时。冷却至室温后,在真空下除去溶剂,并将残余物用ch2cl2萃取。经无水na2so4干燥,然后真空旋干溶剂,粗产物通过硅胶柱色谱法纯化,用二氯甲烷:乙醇(100∶1.5)作为洗脱剂,得到白色固体状产物即为化合物3(1.2g,47.6%)。1hnmr(500mhz,cdcl3,298k):δ8.74–8.71(m,4h,tpy-h3′,5′andtpy-h6,6″),8.67(dt,j=8.0,1.1hz,2h,tpy-h3,3″),7.87(td,j=7.7,1.8hz,2h,tpy-h4,4″),7.82–7.75(m,2h,ph-ha),7.69(d,j=8.1hz,2h,ph-hb),7.52–7.42(m,4h,ph-hc,ph-hd,ph-heandph-hf),7.34(ddd,j=7.5,4.8,1.2hz,2h,tpy-h5,5″),7.31–7.27(m,2h,ph-hg),7.20(d,j=8.1hz,2h,ph-hh),1.33(s,12h,hi).
六齿三联吡啶配体la的合成:
将化合物3(1.5g,2.6mmol),六(4-溴苯基)-苯(328mg,0.33mmol),pd(pph3)2cl2(91mg,0.13mmol)和碳酸钠(1.59g,15mmol)倒入100mlschlenk烧瓶中。在氮气下加入甲苯(30ml),水(15ml)和叔丁醇(7ml)。将混合物在85℃下搅拌6天。冷却至室温后,在真空下除去溶剂,并将残余物用chcl3萃取。合并的有机层用盐水洗涤,经无水na2so4干燥,然后真空浓缩,粗产物通过硅胶柱色谱法纯化,用二氯甲烷:乙醇(100∶3.5)作为洗脱剂,得到白色固体状产物六齿三联吡啶配体la(0.47g,43%)。1hnmr(500mhz,cdcl3,298k)δ8.59(s,12h,tpy-h3′,5′),8.58–8.55(m,12h,tpy-h6,6″),8.51(d,j=8.0hz,12h,tpy-h3,3″),7.74(td,j=7.7,1.8hz,12h,tpy-h4,4″),7.64(d,j=8.1hz,12h,ph-ha),7.43–7.34(m,18h,ph-hc,ph-hdandph-he),7.30(d,j=7.4hz,6h,ph-hf),7.29(s,12h,ph-hj),7.19(ddd,j=7.5,4.7,1.2hz,12h,tpy-h5,5″),7.15(d,j=8.0hz,12h,ph-hb),7.11(d,j=8.0hz,12h,ph-hh),6.99(d,j=8.1hz,12h,ph-hi),6.86(d,j=8.0hz,12h,ph-hg).maldi-tofms(m/z):calcd.for[c240h156n18]+3289.3.found:3289.3
图1是六齿三联吡啶配体la的1hnmr图,图2是la的13cnmr图。通过图3中的2dcosynmr图谱对la的h信号进行了归属。图4的maldi-tof质谱确定了la的分子量为3289.29da。以上表征可充分证明六齿三联吡啶配体la的合成。
向六齿三联吡啶配体la(6.0mg,1.82μmol)的chcl3(2ml)溶液中,添加zn(no3)2·6h2o(1.6mg,5.4μmol)的meoh(6ml)溶液。将混合物在50℃反应8h,然后冷却至室温。加入nh4pf6(80mg)后,静置沉淀并用水洗涤,得到白色产物sa(7.1mg,90%)。反应过程如下:
图5是六螺旋超分子sa的1hnmr图谱,图谱中仅显示出了一套产物峰,表明组装生成了单一的产物。图6的13cnmr图再次证明了组装体sa的形成。通过图7的2dcosynmr图谱对sa的h信号进行了详细的归属。图8的2ddosynmr图谱清楚地显示出一条带(logd=-9.43),再次证实了单一产物的形成。图9是六螺旋超分子sa不同电荷的电喷雾质谱图,图谱中显示了一组分子量为8714.3da的(5+至9+)峰,实验测得的同位素信号能与模拟的同位素信号很好的吻合。图10是六螺旋超分子sa的晶体结构图,可以看到sa由两个平行的六齿配体la和六个zn(ii)组成,并显示出相对拥挤的结构,面板与三联吡啶之间的连接角为63°,面板的直径和高度分别为
实施例2:六螺旋超分子sa对杯[6]芳烃的主客体识别
sa的晶体结构清楚地表明了笼子中有一定的空腔和较大的π共轭面板。此外,三联吡啶的空间位阻为封闭空腔提供了必不可少的帮助,这使得探索客体分子的识别成为可能。为了在室温下促进杯[6]芳烃的溶解,将cd3cn和cdcl3(3:1)的混合物用作溶剂。将sa与杯[6]芳烃混合,超声60min,发现通过添加过量的杯[6]芳烃,sa的氢信号表现出一定的位移,如图11所示,这表示了主客体复合物sa2的形成。图12是主客体复合物sa2不同电荷的电喷雾质谱图,显示了一组分子量为9988.8da的(5+至8+)峰,表示主客体复合物sa2中封装了两个杯[6]芳烃。通过以上表征,可以充分证明六螺旋超分子sa对杯[6]芳烃的主客体识别。
实施例3:三螺旋超分子sb的合成
三齿三联吡啶配体lb的合成路线如下所示,其中化合物1(xiaopengli,j.am.chem.soc.2017,139(24),8174-8185),化合物2(yi-tsuchan,daltontrans.2015,44(11),5139-5145)参照相关文献合成制备,化合物3的合成如实例1所示。
三齿三联吡啶配体lb的合成:
将化合物3(1.2g,2.0mmol),1,3,5-三(4-溴苯基)苯(0.31g,0.57mmol),pd(pph3)2cl2(70mg,0.1mmol)和碳酸钠(1.59g,15mmol)加入到100mlschlenk烧瓶中。在氮气下加入甲苯(30ml),水(15ml)和叔丁醇(5ml)。将混合物在85℃下搅拌3d。冷却至室温后,在真空下除去溶剂,并将残余物用chcl3萃取。合并的有机层用盐水洗涤,经无水na2so4干燥,然后真空浓缩,粗产物通过硅胶柱色谱法纯化,用二氯甲烷:乙醇(100∶2.5)作为洗脱剂,得到白色固体状产物三齿三联吡啶配体lb(0.62g,65%)。1hnmr(500mhz,cdcl3,298k)δ8.73(s,6h,tpy-h3′,5′),8.69(dd,j=4.9,1.7hz,6h,tpy-h6,6″),8.65(d,j=7.9hz,6htpy-h3,3″),7.87–7.79(m,15h,tpy-h4,4″,ph-hkandph-ha),7.75(m,12h,ph-hjandph-hi),7.57(d,j=8.0hz,6h,ph-hh),7.54-7.52(m,6h,ph-hdandph-hc),7.51–7.46(m,6h,ph-hfandph-he),7.35(d,j=8.2hz,6h,ph-hb),7.30(m,12h,tpy-h5,5″andph-hg).maldi-tofms(m/z):calcd.for[c123h81n9]+1683.7.found:1683.7
图13是三齿三联吡啶配体lb的1hnmr图,图14的maldi-tof质谱确定了lb的分子量为1683.66da。以上表征证明了三齿三联吡啶配体lb的合成。
向三齿三联吡啶配体lb(10.0mg,5.9μmol)的chcl3(3ml)溶液中,添加zn(no3)2·6h2o(2.6mg,8.9μmol)的meoh(9ml)溶液。将混合物在50℃反应8h,然后冷却至室温。加入nh4pf6(100mg)后,静置沉淀并用水洗涤,得到白色产物sb(11.8mg,90%)。反应过程如下所示:
图15是三螺旋超分子sb的1hnmr图谱,图谱中仅显示出了一套产物峰,表明组装生成了单一的产物。图16是三螺旋超分子sb不同电荷的电喷雾质谱图,图谱中显示了一组分子量为5637.2da的(3+至6+)峰,实验测得的同位素信号能与模拟的同位素信号很好的吻合。通过以上表征,可证明三螺旋超分子sb的形成。
实施例4:三螺旋超分子sb对杯[4]芳烃和杯[6]芳烃的主客体识别
将三螺旋超分子sb与杯[4]芳烃在乙腈中混合,超声60min,发现通过添加过量的杯[4]芳烃,sb的氢信号表现出一定的位移,如图17所示,这表明了主客体复合物sb2的形成。同样的方法,将sb与过量的杯[6]芳烃在乙腈中混合,超声60min,sb的氢信号表现出一定的位移,如图18所示,这表明了主客体复合物sb3的形成。通过以上核磁表征,可以证明三螺旋超分子sb对杯[4]芳烃和杯[6]芳烃的主客体识别。
- 还没有人留言评论。精彩留言会获得点赞!