一类具有轴手性的联吡啶类配体及其合成方法
一类具有轴手性的联吡啶类配体及其合成方法
1.本发明专利申请是申请日为2014年11月28日、申请号为201410708419.2、发明名称为“一类具有轴手性的联吡啶配体及其合成方法”的中国发明专利申请的分案申请。
技术领域
2.本发明属于有机合成领域,具体涉及一类具有轴手性的联吡啶配体及其合成方法。
背景技术:
3.手性联吡啶配体自1984年首次被合成之后便得到了科研工作者的广泛关注,因此得到了快速发展和应用。手性联吡啶配体能够与多种金属配位用于催化一系列不对称反应,并逐步发展成为不对称催化反应中一类重要的配体,特别是在过渡金属催化的反应中有着重要的应用,因此设计和合成新型手性联吡啶配体仍然是不对称催化研究中的重要内容
1.。(文献1:thummel,r.p;chelucci,g.chem.rev.2002,102,3129.)
4.相比于中心手性和平面手性的联吡啶配体,目前对于轴手性联吡啶配体的研究还相对较少。而已报道的能够用于不对称催化反应中并得到令人满意的结果的联吡啶配体多为中心手性和平面手性的联吡啶配体,由手性骨架传导并控制手性的轴手性联吡啶配体发展极少,而且没有成功应用的实例
2.。(文献2:fletcher,n,c.j.chem.soc.,perkin trans.1,2002,1831.)
5.其实早在1991年,意大利的lucci小组就首次合成了由酒石酸衍生的轴手性联吡啶配体,但是并没有应用于不对称催化反应中
3.。(文献3:bottegi.c.schionato,a.lucci,o,d.synthetic communication 1991,21,1819.)在1996年,milani小组以手性二醇为手性骨架控制联吡啶的轴手性,合成了一种新型轴手性配体,并把该配体应用到一氧化碳与苯乙烯的共聚反应中,但是该配体仅表现出较低的不对称诱导作用
4.。(文献4:milani,b.alessio,e.mestroni,g.zangrando,e.randaccio,l.consiglio,g.j.chem.soc.,dalton trans.,1996,1021.)该小组于2008年又发展了一系列类似的消旋联吡啶配体,将其应用于一氧化碳与苯乙烯的共聚反应,并没有合成相应的手性配体以及尝试不对称催化反应
5.。(文献5:durand,j.zangrando,e.carfagna,c.milani,b.j.chem.soc.,dalton trans.,2008,2171.)
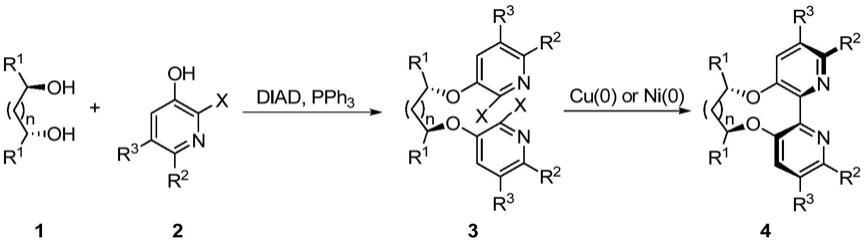
6.从上面的例子中,我们可以看出由手性骨架传导并控制手性的轴手性联吡啶配体发展较少,而且其在不对称反应中的应用也鲜有报道。在基于上述结果,我们设计和合成了一类新的轴手性联吡啶配体。其合成从3
‑
羟基
‑2‑
卤吡啶出发,主要通过mitsunobu反应、镍(0)或铜(0)促进的ullmann反应合成得到。
技术实现要素:
7.本发明的目的是提供一类具有轴手性的联吡啶配体及其合成方法。
8.为实现上述目的,本发明的技术方案如下:
9.一类轴手性联吡啶配体,该配体可以是消旋的或光学活性的,它是下述化合物的结构:
[0010][0011]
其中:
[0012]
r1、r2和r3分别为芳基或c1
‑
c10烷基,所述芳基为苯基或c1
‑
c10的烷基、甲氧基、卤素中一种或二种以上的取代苯基,n为1
‑
5。
[0013]
以化合物3
‑
羟基
‑2‑
卤吡啶为初始原料,通过与手性二醇的mitsunobu反应将吡啶搭载到手性骨架上,随后利用镍(0)或铜(0)促进的ullmann反应实现吡啶的偶联,得到目标联吡啶配体。本发明操作简便,产率高,与传统合成方法相比更为实用。
[0014]
本发明提供的是一类具有轴手性的联吡啶配体的设计和合成,其合成路线如下:
[0015][0016]
其中:
[0017]
x为卤素;
[0018]
n为1
‑
5;
[0019]
r1、r2和r3为芳基或c1
‑
c10烷基,所述芳基为苯基或c1
‑
c10的烷基、甲氧基、卤素中一种或二种以上的取代苯基;
[0020]
cu(0)为铜粉,ni(0)为二价镍化合物被锌粉还原原位生成;
[0021]
反应步骤为:
[0022]
a)氮气保护下,向反应瓶中加入二醇1,吡啶2,三苯基膦,
‑
30
‑
30℃下向该体系加入有机溶剂搅拌溶解,从恒压滴液漏斗中滴加偶氮二甲酸二异丙酯,0
‑
40℃反应10
‑
30小时,得到二吡啶化合物3;其中手性二醇与吡啶化合物的摩尔比为1:2~1:3,手性二醇与三苯基膦的摩尔比为1:2~1:4,手性二醇与偶氮二甲酸二异丙酯的摩尔比为1:2~1:4;
[0023]
b1)铜(0)促进的ullmann偶联:将步骤a得到化合物3,铜粉加入反应瓶中,加入n,n
‑
二甲基甲酰胺,升温至140
‑
180℃反应5
‑
15小时,停止反应;抽滤除去未反应的铜粉,减压蒸馏蒸出n,n
‑
二甲基甲酰胺,有机溶剂溶解,加入强酸搅拌0.5
‑
2小时,后加入碱调节ph至碱性,得目标联吡啶配体4;其中化合物3与铜粉的摩尔比为1:2~1:10;
[0024]
b2)镍(0)促进的ullmann偶联:将二价镍化合物,三苯基膦,n,n
‑
二甲基甲酰胺加入到反应瓶中,搅拌下加入锌粉,0
‑
80℃搅拌0.5
‑
2小时;将步骤a得到的化合物3溶于有机
溶剂中,通过滴液漏斗滴加到反应瓶中,0
‑
80℃反应10
‑
30小时,停止反应;减压蒸馏蒸出n,n
‑
二甲基甲酰胺,所得黑色油状液体用有机溶剂溶解,加入强酸搅拌0.5
‑
2小时,后加入碱调节ph至碱性,得目标联吡啶配体4;其中化合物3与二价镍化合物的摩尔比1:2~1:4,化合物3与锌粉的摩尔比为1:3~1:5,化合物3与三苯基膦的摩尔比为1:8~1:10。
[0025]
所述的有机溶剂为四氢呋喃,乙醚,1,4
‑
二氧六环,二氯甲烷,二氯乙烷,氯仿,苯,甲苯,二甲苯,三甲苯,乙腈,乙酸乙酯,丙酮,甲醇,乙醇中的一种或两种以上的混合。
[0026]
步骤a)所述的吡啶上的卤素x为碘,溴,氯中的一种或两种以上。
[0027]
步骤a)所述的二醇为手性纯试剂,如(2r,4r)
‑
(2,4)
‑
戊二醇和(2r,5r)
‑
(2,5)
‑
己二醇。
[0028]
步骤b2)所述的镍(0)为二价镍化合物被锌粉原位还原。
[0029]
步骤b1)、b2)所述的的是强酸为浓盐酸,硫酸,硝酸中的一种。
[0030]
步骤b1)、b2)所述的碱为氢氧化钠,氢氧化钾,氢化钠,氢化钾,氢化钙,碳酸钠,碳酸钾,碳酸氢钠,碳酸氢钾,碳酸铵,醇钠,醇钾中的一种或两种以上的混合。
[0031]
本发明具有以下优点
[0032]
1.反应步骤少,收率高。
[0033]
2.合成的联吡啶配体无需拆分,为手性纯试剂。
具体实施方式
[0034]
以化合物3
‑
羟基
‑2‑
卤吡啶为初始原料,通过与手性二醇的mitsunobu反应将吡啶搭载到手性骨架上,随后利用镍(0)或铜(0)促进的ullmann反应实现吡啶的偶联,得到目标联吡啶配体。其合成路线如下:
[0035][0036]
其中:
[0037]
x为卤素;
[0038]
n为1
‑
5;
[0039]
r1、r2和r3为芳基或c1
‑
c10烷基,所述芳基为苯基或c1
‑
c10的烷基、甲氧基、卤素中一种或二种以上的取代苯基;
[0040]
cu(0)为铜粉,ni(0)为二价镍化合物被锌粉还原原位生成。
[0041]
下面通过实施例详述本发明;但本发明并不限于下述的实施例。
[0042]
实施例1:联吡啶配体4的合成
[0043][0044]
氮气保护下向250ml反应瓶中加入二醇1(2.080g,20mmol),吡啶2(9.72g,44mmol),三苯基膦(12.6g,48mmol),0℃下加入80ml四氢呋喃搅拌溶解。从恒压滴液漏斗中滴加溶有diad(9.70g,48mmol)的20ml四氢呋喃溶液,滴加完毕,体系呈橙红色透明溶液。反应28小时,原料仅有少量剩余,停止反应。
[0045]
先旋干四氢呋喃,加入20ml二氯甲烷溶解,加入大量石油醚,至有大量固体析出(大部分为三苯氧膦与diad的还原产物。如果无法析出,用小勺反复刮瓶壁),抽滤(抽滤时用石油醚多次洗涤固体,点板检测固体中是否含有目标产物,如果还有较多产物,则将固体再次重结晶),回收母液旋干柱层析得油状液体
‑
化合物3。
[0046]
将化合物3(8.00g,15.7mmol),铜粉(9.98g,157mmol)加入反应瓶中,加入40ml dmf,升温至160℃反应14小时,原料消失。停止反应,抽滤除去未反应的铜粉,减压蒸馏蒸出dmf,所得黑色油状液体用30ml二氯甲烷溶解,加入10ml浓盐酸搅拌2小时,后加入氢氧化钠调节ph至强碱性,有大量固体析出。抽滤,分液,水相用二氯甲烷萃取(20ml
×
3),合并有机相,水洗3次,无水硫酸钠干燥,旋干柱层析得白色固体2.427g。产率=60%。m.p.215
‑
216℃;[α]
20d
=
‑
387.47(c 1.00,chcl3);1h nmr(400mhz,cdcl3)δ8.46(d,j=3.0hz,2h),7.42(dd,j=8.2,1.0hz,2h),7.35
‑
7.26(m,2h),4.73
‑
4.53(m,2h),1.96(s,2h),1.43(d,j=6.5hz,6h);
13
c nmr(100mhz,cdcl3)δ154.2,149.6,144.5,125.0,124.0,75.7,41.9,22.8.hrms:calculated for c
15
h
16
n2o2[m+h]
+
257.1307,found 257.1285。
[0047]
实施例2:联吡啶配体8的合成
[0048][0049]
向四氢呋喃/水(50/50ml)混合溶液中加入吡啶5(6.458g,60mmol),搅拌溶解随后向反应瓶中加入碘(16.8g,66mmol),碳酸氢钠(5.281g,66mmol),室温下反应3天,反应液呈紫黑色,有固体析出,加入10%硫代硫酸钠溶液至紫黑色消失,有大量白色固体析出,抽滤得米白色固体8.19g,经核磁验证为产物6。
[0050]
氮气保护下向250ml反应瓶中加入二醇(2.080g,20mmol),吡啶6(10.3g,44mmol),三苯基膦(12.6g,48mmol),0℃下加入80ml四氢呋喃搅拌溶解。从恒压滴液漏斗中滴加溶有diad(9.70g,48mmol)的20ml四氢呋喃溶液,滴加完毕,体系呈橙红色透明溶液。反应30小时,原料仅有少量剩余,停止反应。旋干柱层析得化合物7粉红色油状液体9.85g。
[0051]
将六水合氯化镍(951mg,4mmol),三苯基膦(4.196g,4mmol)加入到100ml反应瓶中,搅拌下加入锌粉(392mg,6mmol),搅拌1小时,反应液呈红棕色。通过滴液漏斗滴加溶有化合物7的5ml dmf溶液,55℃下反应19小时原料消失,停止反应。减压蒸馏蒸出dmf,所得黑
色油状液体用10ml二氯甲烷溶解,加入2ml浓盐酸搅拌2小时,后加入氢氧化钠调节ph至强碱性。抽滤,分液,水相用二氯甲烷萃取(10ml
×
3),合并有机相,浓缩至10ml左右,加入5ml浓盐酸搅拌2小时,水萃取(10ml
×
4),合并水相,加入氢氧化钠调节ph至强碱性,二氯甲烷萃取(10ml
×
4),干燥旋干柱层析得白色泡沫状固体1.925g。产率=74%。m.p.205
‑
206℃;[α]
20d
=
‑
298.58(c 1.00,chcl3);1h nmr(400mhz,cdcl3)δ7.31(d,j=8.3hz,2h),7.11(d,j=8.3hz,2h),4.64
–
4.47(m,2h),2.59(s,6h),1.91(t,j=4.2hz,2h),1.40(d,j=6.5hz,6h).
13
c nmr(101mhz,cdcl3)δ153.03,152.05,148.66,125.52,123.64,75.67,41.74,24.16,22.76.hrms:calculated for c
17
h
20
n2o2[m+h]
+
285.1598,found 285.1594。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1