一种聚酰亚胺及无色透明聚酰亚胺膜的制作方法
1.本发明涉及柔性显示材料技术领域,尤其涉及一种聚酰亚胺及无色透明聚酰亚胺膜。
背景技术:
2.近年来,聚酰亚胺由于具有优异的耐热性能和可弯曲的柔韧特性,在市场上得到了广泛的应用。尤其是出于器件的轻量化、柔性化的目的,技术人员迫切期望将液晶显示器、oled显示器等图像显示装置中所用的玻璃基板替换为塑料基板,作为最适合作为该塑料材料之一的聚酰亚胺,其研究正在大力进行。
3.然而,传统的聚酰亚胺一般属于全芳香族,且一般由二胺和二酐通过缩聚反应制得预聚物,再经过亚胺化处理制得。由于二胺残基的给电子性和二酐残基的吸电子性导致了分子内电荷的运动,形成电子转移络合物(ctc),从而透光率低,聚酰亚胺膜显示出特征黄色或棕黄色,大大限制了其在图像显示装置中的应用。
4.虽然,国内外学者通过在聚酰亚胺主链上引入含氟基团、脂环结构、非共平面结构、间位取代结构、砜基等抑制ctc的形成,以此来提高聚酰亚胺薄膜的透光性,降低薄膜的黄色指数。但是上述手段的效果有限,还可能会导致薄膜耐热性能的下降。
技术实现要素:
5.基于背景技术存在的技术问题,本发明提出了一种聚酰亚胺及无色透明聚酰亚胺膜,该聚酰胺酰亚胺膜具有非常低的黄色指数的同时还具有优异的耐热尺寸稳定性。
6.本发明提出的一种聚酰亚胺,所述聚酰亚胺是由脂环族二酐、芳香族二酐以及芳香族二酰氯与芳香族二胺进行缩聚后制得;
7.其中,所述脂环族二酐的结构式如下所示:
[0008][0009]
优选地,所述芳香族二酐是4,4'
‑
(六氟异丙烯)二酞酸酐、4,4'
‑
氧双邻苯二甲酸酐、3,3',4,4'
‑
二苯甲酮四甲酸二酐或3,3',4,4'
‑
联苯四羧酸二酐中的至少一种。
[0010]
优选地,所述芳香族二酰氯是对苯二甲酰氯或间苯二甲酰氯中的至少一种。
[0011]
优选地,所述芳香族二胺是2,2'
‑
二(三氟甲基)二氨基联苯、4,4'
‑
二氨基二苯醚或4,4'
‑
二氨基八氟联苯中的至少一种。
[0012]
优选地,所述脂环族二酐的用量是芳香族二胺的30
‑
60mol%,所述芳香族二酰氯的用量是芳香族二胺的10
‑
40mol%。
[0013]
优选地,所述聚酰亚胺具体是由下述方法制备得到:
[0014]
将脂环族二酐、芳香族二酐以及芳香族二酰氯与芳香族二胺在有机溶剂中进行聚
合反应,得到聚酰胺酸;
[0015]
再将上述聚酰胺酸进行亚胺化反应,得到所述聚酰亚胺。
[0016]
优选地,所述有机溶剂是二甲基甲酰胺、二甲基乙酰胺、二甲亚砜、n
‑
甲基吡咯烷酮或二甲苯中的至少一种。
[0017]
本发明还提出一种无色透明聚酰亚胺膜,该聚酰亚胺膜是由上述聚酰亚胺成膜后制得。
[0018]
优选地,所述聚酰亚胺膜的线性热膨胀系数是25ppm/k以下,400nm波长光的透射率为85%以上,黄度指数为3以下。
[0019]
优选地,本发明提出一种图像显示装置,该图像显示装置包括上述聚酰亚胺或聚酰亚胺膜。
[0020]
本发明提供了一种聚酰亚胺及无色透明聚酰亚胺膜,该聚酰亚胺及聚酰亚胺膜通过选择特定结构式的脂环族二酐与芳香族二酐以及芳香族二酰氯复配来制得聚酰亚胺,不仅提高了所制得聚酰亚胺及聚酰亚胺膜的透明度,而且有效降低了所制得聚酰亚胺及聚酰亚胺膜的线热膨胀系数,因此可以广泛用于各种光学元件中。
具体实施方式
[0021]
本发明所提出的聚酰亚胺以及聚酰亚胺膜,是由脂环族二酐、芳香族二酐以及芳香族二酰氯与芳香族二胺进行缩聚后制得;
[0022]
其中,所述脂环族二酐的结构式如下所示:
[0023][0024]
本发明中,脂环族二酐选择为上述结构式所示的二酐,既可以利用其脂肪族骨架结构抑制电荷转移,又可以利用其大分子的桥环烃的结构来阻碍大分子链的密切堆砌。因此上述脂环族二酐的特定选择,使所得聚酰亚胺以及聚酰亚胺膜的高分子链中同时引入了酰胺键、酰亚胺键以及桥环烃的结构,结果不仅提高了对应聚酰亚胺以及聚酰亚胺膜的透光率,降低了黄色指数,同时显著改善了其热膨胀性。
[0025]
本发明中,所述芳香族二酐可选择为4,4'
‑
(六氟异丙烯)二酞酸酐、4,4'
‑
氧双邻苯二甲酸酐、3,3',4,4'
‑
二苯甲酮四甲酸二酐或3,3',4,4'
‑
联苯四羧酸二酐中的至少一种。
[0026]
一方面,芳香族二酐的引入,可与前述脂环族二酐形成复配,使所得聚酰亚胺以及聚酰亚胺膜的高分子链中同时具有芳香族刚性以及脂环族柔性的复合结构,以此确保对应聚酰亚胺以及聚酰亚胺膜的力学性能。另一方面,在考虑到氟取代基的电荷转移效应,将4,4'
‑
(六氟异丙烯)二酞酸酐芳香族二酐作为二酐单体时,显然可进一步改善所得聚酰亚胺以及聚酰亚胺膜的光学透明性能。
[0027]
同样本发明中,所述芳香族二胺可选择为2,2'
‑
二(三氟甲基)二氨基联苯、4,4'
‑
二氨基二苯醚或4,4'
‑
二氨基八氟联苯中的至少一种。同理,当将2,2'
‑
二(三氟甲基)二氨基联苯作为二胺单体时,由于氟取代基的电荷转移效果,可以为所得聚酰亚胺以及聚酰亚
胺膜提供优异的光学透明性能。
[0028]
本发明中,所述芳香族二酰氯可选择为对苯二甲酰氯或间苯二甲酰氯中的至少一种。芳香族二酰氯的引入,与芳香族二胺进行反应,可在所得聚酰亚胺以及聚酰亚胺膜的高分子链中引入酰胺键结构,酰胺键结构增强了高分子链之间的氢键作用力,不仅降低了所得聚酰亚胺以及聚酰亚胺膜的黄色指数,而且对线性热膨胀系数的降低亦有显著改善。
[0029]
本发明中,为了进一步提高所得聚酰亚胺以及聚酰亚胺膜的性能,限定了所述脂环族二酐的用量是芳香族二胺的30
‑
60mol%,所述芳香族二酰氯的用量是芳香族二胺的10
‑
40mol%。
[0030]
上述限定的原料配比可以有效控制所得聚酰亚胺以及聚酰亚胺膜的反应程度以及分子链结构,不仅进一步提高了所得聚酰亚胺以及聚酰亚胺膜的光学透明性能,而且还进一步降低了所得聚酰亚胺以及聚酰亚胺膜的线性热膨胀系数。
[0031]
本发明中,所述聚酰亚胺是通过公知的方法来进行聚合,先得到聚酰亚胺前驱体—聚酰胺酸,再通过热亚胺化法或者化学亚胺化法将聚酰胺酸亚胺化后得到聚酰亚胺。
[0032]
其中,聚酰胺酸是通过将单体加入有机溶剂中,在室温下进行共聚反应得到。该有机溶剂可选择为任意可溶解聚酰胺酸的溶剂,例如为二甲基甲酰胺、二甲基乙酰胺、二甲亚砜、n
‑
甲基吡咯烷酮或二甲苯。
[0033]
本发明中,将聚酰胺酸亚胺化以得到聚酰亚胺优选为通过化学亚胺化法实现。具体是通过向聚酰胺酸溶液中加入脱水剂和酰亚胺化剂,充分反应后完成酰亚胺化反应。该脱水剂可选择为乙酸酐、丙酸酐、马来酸酐或邻苯二甲酸酐等利于反应进行的酸酐类试剂。该酰亚胺化剂可选择为吡啶、异喹啉或三乙胺等叔胺类试剂。
[0034]
本发明中,所述聚酰亚胺膜是通过将上述聚酰亚胺成膜后制得。聚酰亚胺的成膜方法很多,可通过在载体上涂布聚酰亚胺溶液并进行干燥来获得,也可以在载体上涂布聚酰胺酸,先对获得的膜进行加热来完成亚胺化,再进行充分干燥后来获得。
[0035]
上述聚酰亚胺溶液的粘度并无特别限度,要根据涂布厚度及涂布的用途来选择,既不能太低,也不能太高,粘度太低,溶液流动性强,不易成膜,无法保证膜厚的均匀,粘度太高,易出现涂布阻力大,干燥后出现凝胶缺陷等问题。
[0036]
下面,通过具体实施例对本发明的技术方案进行详细说明,但是应该明确提出这些实施例用于举例说明,但是不解释为限制本发明的范围。
[0037]
实施例1
[0038]
一种聚酰亚胺膜,其制备方法包括:
[0039]
在氮气保护下,将3.2023g(10mmol)2,2'
‑
二(三氟甲基)二氨基联苯(tfdb)溶解于50ml的n,n
‑
二甲基乙酰胺中,再依次加入0.8885g(2mmol)4,4'
‑
(六氟异丙烯)二酞酸酐(6fda)、1.9206g(5mmol)降莰烷
‑2‑
螺内酯
‑
α
‑
环戊酮
‑
α'
‑
螺内酯
‑2”‑
降莰烷
‑
5,5”,6,6
”‑
四羧酸二酐(cpoda)以及0.6091g(3mmol)对苯二甲酰氯(tpc),室温下搅拌反应8h,得到聚酰胺酸溶液;
[0040]
向上述聚酰胺酸溶液中加入2.18g吡啶和2.82g乙酸酐,室温下搅拌反应1h后,再升温至70℃下搅拌反应1h,冷却至室温后将反应液移至滴液漏斗中,以2滴/秒的速度滴加至装有1l甲醇的容器中逐渐析出沉淀,将该沉淀过滤后用大量甲醇冲洗,再粉碎所得固体,100℃下真空干燥,得到聚酰亚胺;
[0041]
将上述聚酰亚胺加入n
‑
甲基吡咯烷酮中溶解完全,得到固含量为15wt%的聚酰亚胺溶液,将该聚酰亚胺溶液涂布在玻璃基板上,置于80℃的干燥箱中干燥0.5h,升温至180℃干燥1h,接着升温至260℃干燥0.5h,温度降至室温后取出玻璃板,置于水中脱膜,再将所得薄膜置于干燥箱中干燥除水,即得到聚酰亚胺膜,控制该聚酰亚胺膜的厚度为50μm。
[0042]
上述聚酰亚胺膜的相关性能测试结果如表1中所示。
[0043]
实施例2
[0044]
一种聚酰亚胺膜,其制备方法包括:
[0045]
在氮气保护下,将3.2023g(10mmol)2,2'
‑
二(三氟甲基)二氨基联苯(tfdb)溶解于50ml的n,n
‑
二甲基乙酰胺中,再依次加入0.6204g(2mmol)4,4'
‑
氧双邻苯二甲酸酐(odpa)、1.9206g(5mmol)降莰烷
‑2‑
螺内酯
‑
α
‑
环戊酮
‑
α'
‑
螺内酯
‑2”‑
降莰烷
‑
5,5”,6,6
”‑
四羧酸二酐(cpoda)以及0.6091g(3mmol)对苯二甲酰氯(tpc),室温下搅拌反应8h,得到聚酰胺酸溶液;
[0046]
向上述聚酰胺酸溶液中加入2.18g吡啶和2.82g乙酸酐,室温下搅拌反应1h后,再升温至70℃下搅拌反应1h,冷却至室温后将反应液移至滴液漏斗中,以2滴/秒的速度滴加至装有1l甲醇的容器中逐渐析出沉淀,将该沉淀过滤后用大量甲醇冲洗,再粉碎所得固体,100℃下真空干燥,得到聚酰亚胺;
[0047]
将上述聚酰亚胺加入n
‑
甲基吡咯烷酮中溶解完全,得到固含量为15wt%的聚酰亚胺溶液,将该聚酰亚胺溶液涂布在玻璃基板上,置于80℃的干燥箱中干燥0.5h,升温至180℃干燥1h,接着升温至260℃干燥0.5h,待温度降至室温后取出玻璃板,置于水中脱膜,再将所得薄膜置于干燥箱中干燥除水,即得到聚酰亚胺膜,控制该聚酰亚胺膜的厚度为50μm。
[0048]
上述聚酰亚胺膜的相关性能测试结果如表1中所示。
[0049]
实施例3
[0050]
一种聚酰亚胺膜,其制备方法包括:
[0051]
在氮气保护下,将3.2023g(10mmol)2,2'
‑
二(三氟甲基)二氨基联苯(tfdb)溶解于50ml的n,n
‑
二甲基乙酰胺中,再依次加入0.6445g(2mmol)3,3',4,4'
‑
二苯甲酮四甲酸二酐(btda)、1.9206g(5mmol)降莰烷
‑2‑
螺内酯
‑
α
‑
环戊酮
‑
α'
‑
螺内酯
‑2”‑
降莰烷
‑
5,5”,6,6
”‑
四羧酸二酐(cpoda)以及0.6091g(3mmol)对苯二甲酰氯(tpc),室温下搅拌反应8h,得到聚酰胺酸溶液;
[0052]
向上述聚酰胺酸溶液中加入2.18g吡啶和2.82g乙酸酐,室温下搅拌反应1h后,再升温至70℃下搅拌反应1h,冷却至室温后将反应液移至滴液漏斗中,以2滴/秒的速度滴加至装有1l甲醇的容器中逐渐析出沉淀,将该沉淀过滤后用大量甲醇冲洗,再粉碎所得固体,100℃下真空干燥,得到聚酰亚胺;
[0053]
将上述聚酰亚胺加入n
‑
甲基吡咯烷酮中溶解完全,得到固含量为15wt%的聚酰亚胺溶液,将该聚酰亚胺溶液涂布在玻璃基板上,置于80℃的干燥箱中干燥0.5h,升温至180℃干燥1h,接着升温至260℃干燥0.5h,温度降至室温后取出玻璃板,置于水中脱膜,再将所得薄膜置于干燥箱中干燥除水,即得到聚酰亚胺膜,控制该聚酰亚胺膜的厚度为50μm。
[0054]
上述聚酰亚胺膜的相关性能测试结果如表1中所示。
[0055]
实施例4
[0056]
一种聚酰亚胺膜,其制备方法包括:
[0057]
在氮气保护下,将2.0024g(10mmol)4,4'
‑
二氨基二苯醚(oda)溶解于50ml的n,n
‑
二甲基乙酰胺中,再依次加入0.8885g(2mmol)4,4'
‑
(六氟异丙烯)二酞酸酐(6fda)、1.9206g(5mmol)降莰烷
‑2‑
螺内酯
‑
α
‑
环戊酮
‑
α'
‑
螺内酯
‑2”‑
降莰烷
‑
5,5”,6,6
”‑
四羧酸二酐(cpoda)以及0.6091g(3mmol)对苯二甲酰氯(tpc),室温下搅拌反应8h,得到聚酰胺酸溶液;
[0058]
向上述聚酰胺酸溶液中加入2.18g吡啶和2.82g乙酸酐,室温下搅拌反应1h后,再升温至70℃下搅拌反应1h,冷却至室温后将反应液移至滴液漏斗中,以2滴/秒的速度滴加至装有1l甲醇的容器中逐渐析出沉淀,将该沉淀过滤后用大量甲醇冲洗,再粉碎所得固体,100℃下真空干燥,得到聚酰亚胺;
[0059]
将上述聚酰亚胺加入n
‑
甲基吡咯烷酮中溶解完全,得到固含量为15wt%的聚酰亚胺溶液,将该聚酰亚胺溶液涂布在玻璃基板上,置于80℃的干燥箱中干燥0.5h,升温至180℃干燥1h,接着升温至260℃干燥0.5h,待温度降至室温后取出玻璃板,置于水中脱膜,再将所得薄膜置于干燥箱中干燥除水,即得到聚酰亚胺膜,控制该聚酰亚胺膜的薄膜厚度为50μm。
[0060]
上述聚酰亚胺膜的相关性能测试结果如表1中所示。
[0061]
实施例5
[0062]
一种聚酰亚胺膜,其制备方法包括:
[0063]
在氮气保护下,将3.2023g(10mmol)2,2'
‑
二(三氟甲基)二氨基联苯(tfdb)溶解于50ml的n,n
‑
二甲基乙酰胺中,再依次加入0.8885g(2mmol)4,4'
‑
(六氟异丙烯)二酞酸酐(6fda)、1.9206g(5mmol)降莰烷
‑2‑
螺内酯
‑
α
‑
环戊酮
‑
α'
‑
螺内酯
‑2”‑
降莰烷
‑
5,5”,6,6
”‑
四羧酸二酐(cpoda)以及0.6091g(3mmol)间苯二甲酰氯(ipc),室温下搅拌反应8h,得到聚酰胺酸溶液;
[0064]
向上述聚酰胺酸溶液中加入2.18g吡啶和2.82g乙酸酐,在室温下搅拌反应1h后,再升温至70℃下搅拌反应1h,冷却至室温后将反应液移至滴液漏斗中,以2滴/秒的速度滴加至装有1l甲醇的容器中逐渐析出沉淀,将该沉淀过滤后用大量甲醇冲洗,再粉碎所得固体,100℃下真空干燥,得到聚酰亚胺;
[0065]
将上述聚酰亚胺加入n
‑
甲基吡咯烷酮中溶解完全,得到固含量为15wt%的聚酰亚胺溶液,将该聚酰亚胺溶液涂布在玻璃基板上,置于80℃的干燥箱中干燥0.5h,升温至180℃干燥1h,接着升温至260℃干燥0.5h,待温度降至室温后取出玻璃板,置于水中脱膜,再将所得薄膜置于干燥箱中干燥除水,即得到聚酰亚胺膜,控制该聚酰亚胺膜的厚度为50μm。
[0066]
上述聚酰亚胺膜的相关性能测试结果如表1中所示。
[0067]
实施例6
[0068]
一种聚酰亚胺膜,其制备方法包括:
[0069]
在氮气保护下,将3.2023g(10mmol)2,2'
‑
二(三氟甲基)二氨基联苯(tfdb)溶解于50ml的n,n
‑
二甲基乙酰胺中,再依次加入1.3327g(3mmol)4,4'
‑
(六氟异丙烯)二酞酸酐(6fda)、1.1524g(3mmol)降莰烷
‑2‑
螺内酯
‑
α
‑
环戊酮
‑
α'
‑
螺内酯
‑2”‑
降莰烷
‑
5,5”,6,6
”‑
四羧酸二酐(cpoda)以及0.8121g(4mmol)对苯二甲酰氯(tpc),室温下搅拌反应8h,得到聚酰胺酸溶液;
[0070]
向上述聚酰胺酸溶液中加入2.18g吡啶和2.82g乙酸酐,在室温下搅拌1h后,再升
温至70℃下搅拌反应1h,冷却至室温后将反应液移至滴液漏斗中,以2滴/秒的速度滴加至装有1l甲醇的容器中逐渐析出沉淀,将该沉淀过滤后用大量甲醇冲洗,再粉碎所得固体,100℃下真空干燥,得到聚酰亚胺;
[0071]
将上述聚酰亚胺加入n
‑
甲基吡咯烷酮中溶解完全,得到固含量为15wt%的聚酰亚胺溶液,将该聚酰亚胺溶液涂布在玻璃基板上,置于80℃的干燥箱中干燥0.5h,升温至180℃干燥1h,接着升温至260℃干燥0.5h,待温度降至室温后取出玻璃板,置于水中脱膜,再将所得薄膜置于干燥箱中干燥除水,即得到聚酰亚胺膜,控制该聚酰亚胺膜的厚度为50μm。
[0072]
上述聚酰亚胺膜的相关性能测试结果如表1中所示。
[0073]
实施例7
[0074]
一种聚酰亚胺膜,其制备方法包括:
[0075]
在氮气保护下,将3.2023g(10mmol)2,2'
‑
二(三氟甲基)二氨基联苯(tfdb)溶解于50ml的n,n
‑
二甲基乙酰胺中,再依次加入1.3327g(3mmol)4,4'
‑
(六氟异丙烯)二酞酸酐(6fda)、2.3047g(6mmol)降莰烷
‑2‑
螺内酯
‑
α
‑
环戊酮
‑
α'
‑
螺内酯
‑2”‑
降莰烷
‑
5,5”,6,6
”‑
四羧酸二酐(cpoda)以及0.2030g(1mmol)对苯二甲酰氯(tpc),室温下搅拌反应8h,得到聚酰胺酸溶液;
[0076]
向上述聚酰胺酸溶液中加入2.18g吡啶和2.82g乙酸酐,在室温下搅拌反应1h后,再升温至70℃下搅拌反应1h,冷却至室温后将反应液移至滴液漏斗中,以2滴/秒的速度滴加至装有1l甲醇的容器中逐渐析出沉淀,将该沉淀过滤后用大量甲醇冲洗,再粉碎所得固体,100℃下真空干燥,得到聚酰亚胺;
[0077]
将上述聚酰亚胺加入n
‑
甲基吡咯烷酮中溶解完全,得到固含量为15wt%的聚酰亚胺溶液,将该聚酰亚胺溶液涂布在玻璃基板上,置于80℃的干燥箱中干燥0.5h,升温至180℃干燥1h,接着升温至260℃干燥0.5h,温度降至室温后取出玻璃板,置于水中脱膜,再将所得薄膜置于干燥箱中干燥除水,即得到聚酰亚胺膜,控制该聚酰亚胺膜的厚度为50μm。
[0078]
上述聚酰亚胺膜的相关性能测试结果如表1中所示。
[0079]
对比例1
[0080]
一种聚酰亚胺膜,其制备方法包括:
[0081]
在氮气保护下,将3.2023g(10mmol)2,2'
‑
二(三氟甲基)二氨基联苯(tfdb)溶解于50ml的n,n
‑
二甲基乙酰胺中,再依次加入0.8885g(2mmol)4,4'
‑
(六氟异丙烯)二酞酸酐(6fda)、0.9806g(5mmol)环丁烷四甲酸二酐(cbda)以及0.6091g(3mmol)对苯二甲酰氯(tpc),室温下搅拌反应8h,得到聚酰胺酸溶液;
[0082]
向上述聚酰胺酸溶液中加入2.18g吡啶和2.82g乙酸酐,室温下搅拌反应1h后,再升温至70℃下搅拌反应1h,冷却至室温后将反应液移至滴液漏斗中,以2滴/秒的速度滴加至装有1l甲醇的容器中逐渐析出沉淀,将该沉淀过滤后用大量甲醇冲洗,再粉碎所得固体,100℃下真空干燥,得到聚酰亚胺;
[0083]
将上述聚酰亚胺加入n
‑
甲基吡咯烷酮中溶解完全,得到固含量为15wt%的聚酰亚胺溶液,将该聚酰亚胺溶液涂布在玻璃基板上,置于80℃的干燥箱中干燥0.5h,升温至180℃干燥1h,接着升温至260℃干燥0.5h,温度降至室温后取出玻璃板,置于水中脱膜,再将所得薄膜置于干燥箱中干燥除水,即得到聚酰亚胺膜,控制该聚酰亚胺膜的厚度为50μm。
[0084]
上述聚酰亚胺膜的相关性能测试结果如表1中所示。
[0085]
对比例2
[0086]
一种聚酰亚胺膜,其制备方法包括:
[0087]
在氮气保护下,将3.2023g(10mmol)2,2'
‑
二(三氟甲基)二氨基联苯(tfdb)溶解于50ml的n,n
‑
二甲基乙酰胺中,再依次加入1.3327g(3mmol)4,4'
‑
(六氟异丙烯)二酞酸酐(6fda)、0.7682g(2mmol)降莰烷
‑2‑
螺内酯
‑
α
‑
环戊酮
‑
α'
‑
螺内酯
‑2”‑
降莰烷
‑
5,5”,6,6
”‑
四羧酸二酐(cpoda)以及1.0151g(5mmol)对苯二甲酰氯(tpc),室温下搅拌反应8h,得到聚酰胺酸溶液;
[0088]
向上述聚酰胺酸溶液中加入2.18g吡啶和2.82g乙酸酐,室温下搅拌反应1h后,再升温至70℃下搅拌反应1h,冷却至室温后将反应液移至滴液漏斗中,以2滴/秒的速度滴加至装有1l甲醇的容器中逐渐析出沉淀,将该沉淀过滤后用大量甲醇冲洗,再粉碎所得固体,100℃下真空干燥,得到聚酰亚胺;
[0089]
将上述聚酰亚胺加入n
‑
甲基吡咯烷酮中溶解完全,得到固含量为15wt%的聚酰亚胺溶液,将该聚酰亚胺溶液涂布在玻璃基板上,置于80℃的干燥箱中干燥0.5h,升温至180℃干燥1h,接着升温至260℃干燥0.5h,待温度降至室温后取出玻璃板,置于水中脱膜,再将所得薄膜置于100℃干燥箱中干燥除水,即得到聚酰亚胺膜,控制该聚酰亚胺膜的厚度为50μm。
[0090]
上述聚酰亚胺膜的相关性能测试结果如表1中所示。
[0091]
对比例3
[0092]
一种聚酰亚胺膜,其制备方法包括:
[0093]
在氮气保护下,将3.2023g(10mmol)2,2'
‑
二(三氟甲基)二氨基联苯(tfdb)溶解于50ml的n,n
‑
二甲基乙酰胺中,再依次加入0.4442g(1mmol)4,4'
‑
(六氟异丙烯)二酞酸酐(6fda)、2.6888g(7mmol)降莰烷
‑2‑
螺内酯
‑
α
‑
环戊酮
‑
α'
‑
螺内酯
‑2”‑
降莰烷
‑
5,5”,6,6
”‑
四羧酸二酐(cpoda)以及0.4060g(2mmol)对苯二甲酰氯(tpc),室温下搅拌反应8h,得到聚酰胺酸溶液;
[0094]
向上述聚酰胺酸溶液中加入2.18g吡啶和2.82g乙酸酐,室温下搅拌反应1h后,再升温至70℃下搅拌反应1h,冷却至室温后将反应液移至滴液漏斗中,以2滴/秒的速度滴加至装有1l甲醇的容器中逐渐析出沉淀,将该沉淀过滤后用大量甲醇冲洗,再粉碎所得固体,100℃下真空干燥,得到聚酰亚胺;
[0095]
将上述聚酰亚胺加入n
‑
甲基吡咯烷酮中溶解完全,得到固含量为15wt%的聚酰亚胺溶液,将该聚酰亚胺溶液涂布在玻璃基板上,置于80℃的干燥箱中干燥0.5h,升温至180℃干燥1h,接着升温至260℃干燥0.5h,温度降至室温后取出玻璃板,置于水中脱膜,再将所得薄膜置于干燥箱中干燥除水,即得到聚酰亚胺膜,控制该聚酰亚胺膜的厚度为50μm。
[0096]
上述聚酰亚胺膜的相关性能测试结果如表1中所示。
[0097]
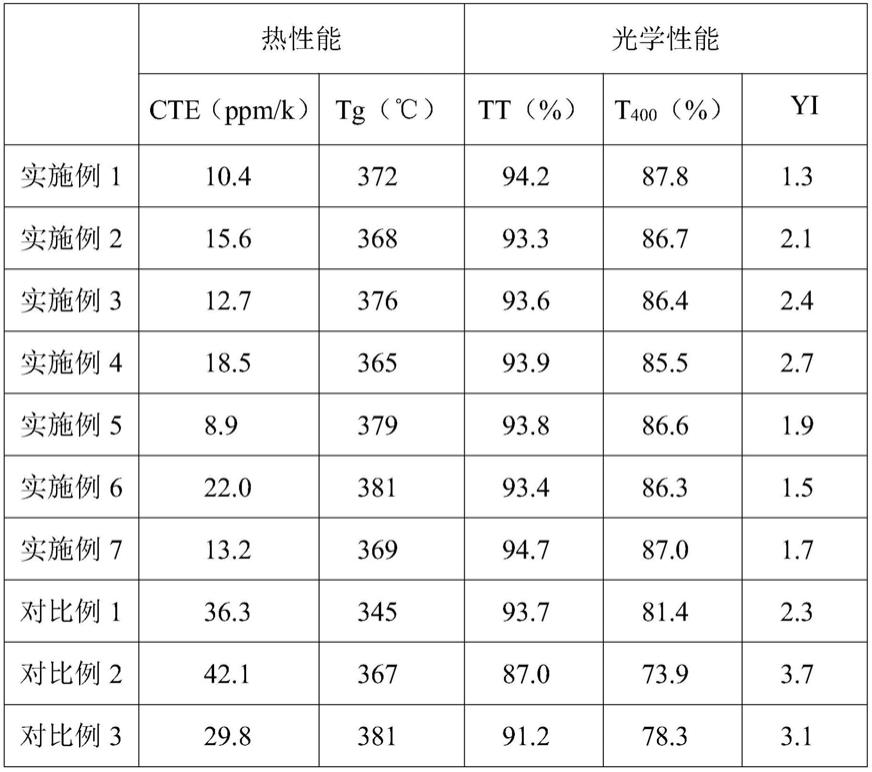
将实施例1
‑
7和对比例1
‑
3所得聚酰亚胺膜的热性能以及光学性能的测试结果在下表中示出。
[0098]
热性能包括玻璃化转变温度和线性热膨胀系数:
[0099]
线性热膨胀系数cte:使用热机械式分析装置,在氮气氛围下,施加50mn的荷重,以10℃/min的升温速度在50
‑
250℃的温度范围内进行测定,求出平均值;
[0100]
玻璃化转变温度tg:使用差式扫描热量计装置,在氮气氛围下,以10℃/min的升温
速度在50
‑
450℃的温度范围内进行dsc测定,求出玻璃化转变温度。
[0101]
光学性能包括透光率和黄度指数:
[0102]
总光线透射率tt:使用紫外
‑
可见光谱测定总光线透射率;
[0103]
400nm光处测定透射率t
400
:使用紫外分光光度计测定400nm光处透射率;
[0104]
黄度指数yi:使用紫外分光光度计,根据astm e313标准测定黄度指数。
[0105]
表1实施例和对比例所得聚酰亚胺膜的测试结果
[0106][0107]
从上表可以看出,本发明实施例所述聚酰亚胺膜在短波长400nm区域的整显示出高的透光率,并且具有低的黄度指数和线性热膨胀系数。
[0108]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1