一种LS007杂质化合物A及其制备工艺和应用的制作方法
一种ls007杂质化合物a及其制备工艺和应用
技术领域
1.本发明属于化学制药技术领域,具体涉及一种ls007杂质化合物a及其制备工艺、原料配方、应用。
背景技术:
2.增殖性疾病如癌症的特征是不受控的和失调的细胞增殖。蛋白激酶是癌症研究的一类重要的酶。蛋白激酶家族是人类基因组中最大的家族之一,大多数激酶都有保守的功能结构域,由250-300个氨基酸残基构成催化结构域,该结构域包含atp的结合位点,atp的磷酸基团以共价方式被转移至底物分子,使底物磷酸化。蛋白激酶可根据作用的底物类型进行分类,例如丝氨酸激酶、苏氨酸激酶、酪氨酸激酶等。
3.激酶是催化磷酸基团从含有磷酸的高能分子转移到特定底物的酶,介导细胞内信号通路激活,多种细胞外刺激和其它刺激,作为可调整或调节靶蛋白质生物学功能的分子开关作用,触发激酶使底物磷酸化。细胞外刺激可影响一种或多种与细胞生长、迁移、分化、激素分泌、转录因子活化、肌肉收缩、葡萄糖代谢、蛋白质合成控制和细胞周期调节等有关的细胞应答。若激酶介导异常细胞应答,将导致多种疾病,包括但不限于变态反应和哮喘、阿尔茨海默病、自身免疫疾病、骨疾病、癌症、心血管疾病、炎性疾病、激素相关性疾病、代谢疾病、神经学疾病和神经变性疾病等。
4.现有技术中已知众多能够通过阻断atp结合来抑制蛋白激酶功能的分子。细胞周期蛋白依赖性激酶(cdk)是与多种细胞周期蛋白亚单位相关的丝氨酸/苏氨酸蛋白激酶,在调节细胞周期过程和转录周期中起关键作用。在真核细胞中的多种重要调节途径,包括细胞周期控制、细胞凋亡、神经元生理学、分化和转录中牵涉10种不同的cdk(cdk1-9和11)。)。另外,病毒的复制过程也需要cdks,特别是cdk2、cdk7和cdk9。已有报道显示,限制病毒复制的cdks抑制剂,能作用于许多病毒,包括人免疫缺陷病毒、人巨细胞病毒、疱疹病毒和水痘-带状疱疹病毒。
5.其中,cdk9的抑制剂是目前治疗心血管疾病、包括心脏肥大的潜在治疗的一种新策略。心脏肥大的特征是mrna和蛋白质合成的总量增加,cdk7和cdk9是转录的主要驱动因子,与心脏肥大密切相关。因此,抑制cdk9及其相关细胞周期蛋白是心血管疾病的有效治疗策略。
6.cdks的抑制剂还可用于治疗神经变性紊乱的疾病,如阿尔茨海默病。与阿尔茨海默病有关的双股螺旋形细丝(paired helical filaments)的出现是由于cdk5/p25使tau蛋白高度磷酸化而引起的。
7.公开号为cn103373994a(整体援引加入本文)的中国发明专利公开了一类具有cdk-9抑制功能的化合物及其制备方法;公开号为cn108658966a的中国发明专利公开了化合物3-(5-氟-4-(4-甲基-2-(甲基氨基)噻唑-5-基)嘧啶-2-基氨基)-苯磺酰胺(以下简称化合物ls007)的酒石酸盐和晶型,其具有良好的性能,具有开发成为选择性cdk9激酶抑制剂的潜力。
8.但,目前针对化合物3-(5-氟-4-(4-甲基-2-(甲基氨基)噻唑-5-基)嘧啶-2-基氨基)-苯磺酰胺合成过程中的杂质及其质量控制研究仍然是空白。
技术实现要素:
9.本发明提供了一种ls007杂质化合物a及其制备工艺和应用。
10.为了解决上述技术问题,本发明提供了一种化合物(即ls007杂质化合物a)的制备工艺,包括:以物质a1-5、盐酸、亚硝酸钠溶液为主原料进行第六反应;其中所述物质a1-5的结构式为
11.第二方面,本发明提供了一种化合物,所述化合物的结构式为
12.第三方面,本发明提供了一种如前所述的化合物在作为药用化合物分析用标准品的应用。
13.本发明的有益效果是,首次提供了一种化合物及其合成方法,该化合物为ls007合成过程中的主要杂质之一,对ls007的质量控制具有重要意义。本发明的化合物以物质a1-5、盐酸、亚硝酸钠溶液为主原料进行第六反应制备所述化合物,可以用于药用化合物分析的标准品。
14.本发明的其他特征和优点将在随后的说明书中阐述,并且,部分地从说明书中变得显而易见,或者通过实施本发明而了解。本发明的目的和其他优点在说明书、权利要求书以及附图中所特别指出的结构来实现和获得。
15.为使本发明的上述目的、特征和优点能更明显易懂,下文特举较佳实施例,并配合所附附图,作详细说明如下。
附图说明
16.为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
17.图1是本发明的化合物的制备工艺流程图;
18.图2是本发明的物质a1-5的制备工艺流程图;
19.图3是本发明的物质a1-4的制备工艺流程图;
20.图4是本发明的物质a1-3的制备工艺流程图;
21.图5是本发明的物质a1-2的制备工艺流程图;
22.图6是本发明的物质a1-1的制备工艺流程图;
23.图7是本发明的化合物的红外光谱图;
24.图8是本发明的化合物的核磁共振氢谱图;
25.图9是本发明的化合物的核磁共振碳谱图;
26.图10是本发明的化合物的质谱图;
27.图11是将本发明合成得到的杂质化合物a、化合物a1-3作为标准对照品加入ls007后的系统适用性hplc色谱图;
28.图12是按照现有技术合成得到的ls007的hplc色谱图。
具体实施方式
29.为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合附图对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
30.第一部分
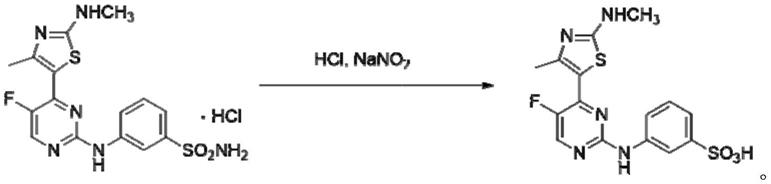
31.现有技术的缺点,为此,见图1,本发明提供了一种化合物的制备工艺,包括:以物质a1-5、盐酸、亚硝酸钠溶液为主原料进行第六反应;其中所述物质a1-5的结构式为
[0032][0033]
可选的,见图1,所述第六反应包括:将物质a1-5、盐酸在冰浴下滴加亚硝酸钠溶液,并在室温下进行反应;将反应液缓慢倒入水中,析出固体;搅拌并滤取所述固体;以及烘干,得到所述化合物。具体的,在烧瓶中加入物质a1-5、盐酸,在冰浴下滴加亚硝酸钠溶液,滴加完室温反应1h,将反应液缓慢倒入水中,析出黄色固体,搅拌1h,过滤,烘干,得到所述化合物。
[0034]
所述第六反应的反应式为
[0035][0036]
作为物质a1-5的制备方法的一种可选实施方式。
[0037]
见图2,所述物质a1-5的制备方法包括:以物质a1-3、物质a1-4、乙二醇单甲醚为主
原料进行第五反应;其中所述物质a1-3的结构式为所述物质a1-4的结构式为所述第五反应包括:粗制和精制;其中所述粗制包括:将所述物质a1-3、所述物质a1-4、乙二醇单甲醚混合搅拌;升温至124~126℃,在回流体系下进行反应;将反应液降温至60℃以下,并转移至冰水中;室温下搅拌;抽滤,得到滤饼;将所述滤饼用水淋洗,得到粗品;所述精制包括:将所述粗品加入乙二醇单甲醚中,并在25
±
5℃下搅拌;抽滤,得到滤饼;将所述滤饼用乙二醇单甲醚淋洗;在50
±
5℃干燥,得到所述物质a1-5;所述第五反应的反应式为:
[0038][0039]
具体的,粗制:在反应釜中投入物质a1-3和物质a1-4,加入乙二醇单甲醚,搅拌,升温,于124~126℃(体系回流)下反应22
±
2小时;降温使反应混合液温度降至60℃以下,然后将料液慢慢转移至22l的冰水中,室温搅拌约0.5小时;抽滤,滤饼用少量水淋洗,得到粗品。精制:在四口瓶中投入上述粗品,加入乙二醇单甲醚,25
±
5℃下搅拌2小时,抽滤,滤饼用适量乙二醇单甲醚淋洗,所得滤饼热风干燥(50
±
5℃),即得到棕色固体,即所述物质a1-5。可选的,在第五反应中,物质a1-3、物质a1-4的摩尔比为1:1.05~1.2,优选为1:1.1。
[0040]
作为物质a1-4的制备方法的一种可选实施方式。
[0041]
见图3,所述物质a1-4的制备方法包括:以间氨基苯磺酰胺、单氰氨、乙腈为主原料进行第四反应;所述第四反应包括:将间氨基苯磺酰胺、单氰氨、乙腈混合搅拌;降温至5
±
1℃并滴加三甲基氯硅烷;升温至回流,并保温进行反应;将反应混合物冷至室温;抽滤,得到滤饼;将所述滤饼用乙腈淋洗;以及60
±
2℃烘干,得到所述物质a1-4;所述第四反应的反应式为具体的,在反应瓶中投入间氨基苯磺酰胺、单氰氨、乙腈,搅拌;降温至5
±
1℃,滴加三甲基氯硅烷,滴加完毕后,升温至回流,保温反应30
±
1小时;反应混合物冷至室温,抽滤得到滤饼,少量乙腈淋洗,60
±
2℃热风中烘干,得到所述物质a1-4。可选的,在第四反应中,所述间氨基苯磺酰胺、单氰胺、三甲基氯硅烷的摩尔比物质a1-3、物质a1-4的摩尔比为1:1.05~1.2:1.05~1.2,优选为1:1.1:1.1。
[0042]
作为物质a1-3的制备方法的一种可选实施方式。
[0043]
见图4,所述物质a1-3的制备方法包括:以物质a1-2、甲醇、二氯甲烷、乙腈、氟试剂
为主原料进行第三反应;其中所述物质a1-2的结构式为所述第三反应包括:将物质a1-2、甲醇、二氯甲烷、乙腈混合搅拌;冷至-5
±
1℃,并加入氟试剂selectfluor进行反应;加入氨甲醇溶液(浓度为8mol/l)以猝灭反应,继续搅拌;30℃下旋蒸;加入水,并搅拌过滤;室温下风干,得到结晶性固体;以及柱层析纯化,得到所述物质a1-3;所述第三反应的反应式为具体的,在反应釜中投入物质a1-2、甲醇、二氯甲烷、乙腈,搅拌,冷至-5
±
1℃,加入氟试剂,反应0.5小时,加入氨甲醇溶液,继续搅拌过夜,30℃下旋蒸溶剂,加入水,搅拌30分钟后过滤,室温条件下风干,得到黄色结晶性固体,柱层析纯化得到所述物质a1-3。可选的,在第三反应中,所述a1-2与氟试剂、氨甲醇的摩尔比为1:1~1.5:1~5,优选为1:1~1.5:1~3;更进一步的是1:1:2。
[0044]
作为物质a1-2的制备方法的一种可选实施方式。
[0045]
见图5,所述物质a1-2的制备方法包括:以物质a1-1、n,n-二甲基甲酰胺二甲基缩醛、二甲苯为主原料进行第二反应;其中所述物质a1-1的结构式为所述第二反应包括:将物质a1-1、n,n-二甲基甲酰胺二甲基缩醛、二甲苯混合搅拌;升温至回流,并保温进行反应;减压蒸除溶液,得到粘稠固体;降温至60℃,并在所述粘稠固体中加入甲醇;加热至回流;降温析晶;过滤,得到滤饼;将所述滤饼用甲醇淋洗;60
±
2℃烘干,得到所述物质a1-2;所述第二反应的反应式为具体的,在反应瓶中投入物质a1-1、n,n-二甲基甲酰胺二甲基缩醛、二甲苯,搅拌,升温至回流,保温反应24
±
1小时;反应结束后,减压蒸除溶液得橙色粘稠固体,降温至60℃加入甲醇,后加热至回流1小时,降温析晶,过滤,滤饼用少量甲醇淋洗,60
±
2℃热风中烘干,得到所述物质a1-2。可选的,在第二反应中,所述物质a1-1、n,n-二甲基甲酰胺二甲基缩醛的摩尔比为1:(1.5~3),优选为1:1.9。
[0046]
作为物质a1-1的制备方法的一种可选实施方式。
[0047]
见图6,所述物质a1-1的制备方法包括:以n-甲基硫脲、甲醇、吡啶、3-氯-2,4-戊二酮为主原料进行第一反应;其中所述第一反应包括:将n-甲基硫脲、甲醇混合搅拌;加入吡啶,呈现白色悬浊液;降温至0~5℃,并滴加3-氯-2,4-戊二酮;30~35℃保温并进行反应;将反应液倒入水中,继续搅拌;抽滤,得到滤饼;将所述滤饼用水淋洗;在80
±
2℃烘干,得到
2,4-戊二酮(分子量134.6,1.68mol)。滴加完毕后,30~35℃保温反应2小时,将反应液倒入3l水中,继续搅拌半小时,抽滤,水淋洗滤饼3次,滤饼在80
±
2℃热风烘干,得到166g物质a1-1(分子量170.2,0.976mol),纯度99%,收率86.3%。
[0059]
(2)制备物质a1-2
[0060]
在1l反应瓶中,投入161.8g物质a1-1(分子量170.2,0.95mol)、333.8g n,n-二甲基甲酰胺二甲基缩醛(分子量119.2,2.8mol)、220g二甲苯,搅拌,升温至回流,保温反应24
±
1小时。反应结束后,减压蒸除溶液得橙色粘稠固体,降温至60℃加入220ml甲醇,后加热至回流1小时,降温析晶,过滤,滤饼用少量甲醇淋洗,60
±
2℃热风中烘干,得123.9g物质a1-2(分子量225.3,0.55mol),纯度98%,收率57%。
[0061]
(3)制备物质a1-3
[0062]
在3l反应釜中,投入56.8g物质a1-2(分子量225.3,0.25mol)、450ml甲醇、680ml二氯甲烷、450ml乙腈,搅拌,冷至-5
±
1℃,加入131.1g氟试剂(分子量354.3,0.37mol),反应0.5小时,加入氨甲醇溶液63.3g(氨浓度为8mol/l),继续搅拌过夜,30℃下旋蒸溶剂,加入500g水,搅拌30分钟后过滤,室温条件下风干,得到黄色结晶性固体,柱层析纯化,得到34.1g物质a1-3(分子量243.3,0.140mol),纯度95%,收率53%。
[0063]
(4)制备物质a1-4
[0064]
在1l反应瓶中,投入67.5g间氨基苯磺酰胺(分子量172.2,0.39mol)、19.7g单氰胺(分子量42.0,0.47mol)、660ml乙腈,搅拌。降温至5
±
1℃,滴加51.0g三甲基氯硅烷(分子量108.6,0.47mol),滴加完毕后,升温至回流,保温反应30
±
1小时。反应混合物冷至室温,抽滤得到滤饼,少量乙腈淋洗,60
±
2℃热风中烘干,得78.6g物质a1-4(分子量250.7,0.33mol),纯度95%,收率80%。
[0065]
(5)制备物质a1-5
[0066]
粗制:在反应釜中,投入10g物质a1-3(分子量243.3,0.041mol)和12.3g物质a1-4分子量250.7,0.049mol),加入100g乙二醇单甲醚(分子量76.1,1.31mol),搅拌,升温,于124~126℃(体系回流)下反应22
±
2小时;降温使反应混合液温度降至60℃以下,然后将料液慢慢转移至22l的冰水中,室温搅拌约0.5小时;抽滤,滤饼用少量水淋洗,得到粗品。
[0067]
精制:在四口瓶中,投入上述粗品,加入18g乙二醇单甲醚,25
±
5℃下搅拌2小时,抽滤,滤饼用适量乙二醇单甲醚淋洗,所得滤饼热风干燥(50
±
5℃),得到12.1g棕色固体,即物质a1-5(分子量430.9,0.028mol),纯度98%,收率67%。
[0068]
(6)制备化合物
[0069]
在烧瓶中加入10g物质a1-5(分子量430.9,0.023mol)、0.2l盐酸,冰浴下滴加10g亚硝酸钠的水溶液(含有亚硝酸钠4g,0.058mol),滴加完室温反应1h,将反应液缓慢倒入1l水中,析出黄色固体,搅拌1h,过滤烘干,得4.2g化合物(分子量395.4,0.011mol),纯度98%,收率47.8%。
[0070]
实施例2
[0071]
(1)制备物质a1-1
[0072]
在1l反应瓶中加入100.5g n-甲基硫脲(分子量90.1,1.12mol)、477g甲醇,搅拌,加入88.2g吡啶(分子量79.1,1.12mol),呈现白色悬浊液。降温至0~5℃,滴加150g 3-氯-2,4-戊二酮(分子量134.6,1.11mol)。滴加完毕后,30~35℃保温反应2小时,将反应液倒入
2l水中,继续搅拌半小时,抽滤,水淋洗滤饼3次,滤饼在80
±
2℃热风烘干,得到164g物质a1-1(分子量170.2,0.964mol),纯度99%,收率86.0%。
[0073]
(2)制备物质a1-2
[0074]
在1l反应瓶中,投入161.8g物质a1-1(分子量170.2,0.95mol)、214.9g n,n-二甲基甲酰胺二甲基缩醛(分子量119.2,1.8mol)、176.3g二甲苯,搅拌,升温至回流,保温反应24
±
1小时。反应结束后,减压蒸除溶液得橙色粘稠固体,降温至60℃加入200ml甲醇,后加热至回流1小时,降温析晶,过滤,滤饼用少量甲醇淋洗,60
±
2℃热风中烘干,得114.7g物质a1-2(分子量225.3,0.51mol),纯度98%,收率52%。
[0075]
(3)制备物质a1-3
[0076]
在3l反应釜中,投入56.8g物质a1-2(分子量225.3,0.25mol)、430ml甲醇、648ml二氯甲烷、430ml乙腈,搅拌,冷至-5
±
1℃,加入89.4g氟试剂(分子量354.3,0.25mol),反应0.5小时,加入氨甲醇溶液63.3g(氨浓度为8mol/l),继续搅拌过夜,30℃下旋蒸溶剂,加入500g水,搅拌30分钟后过滤,室温条件下风干,得到黄色结晶性固体,柱层析纯化,得到32.1g物质a1-3(分子量243.3,0.132mol),纯度95%,收率50%。
[0077]
(4)制备物质a1-4
[0078]
在1l反应瓶中,投入67.5g间氨基苯磺酰胺(分子量172.2,0.39mol)、18.1g单氰胺(分子量42.0,0.43mol)、600ml乙腈,搅拌。降温至5
±
1℃,滴加46.7g三甲基氯硅烷(分子量108.6,0.43mol),滴加完毕后,升温至回流,保温反应30
±
1小时。反应混合物冷至室温,抽滤得到滤饼,少量乙腈淋洗,60
±
2℃热风中烘干,得77g物质a1-4(分子量250.7,0.31mol),纯度95%,收率75%。
[0079]
(5)制备物质a1-5
[0080]
粗制:在反应釜中,投入10g物质a1-3(分子量243.3,0.041mol)和11.3g物质a1-4分子量250.7,0.045mol),加入93.4g乙二醇单甲醚(分子量76.1,1.23mol),搅拌,升温,于124~126℃(体系回流)下反应22
±
2小时;降温使反应混合液温度降至60℃以下,然后将料液慢慢转移至22 l的冰水中,室温搅拌约0.5小时;抽滤,滤饼用少量水淋洗,得到粗品。
[0081]
精制:在四口瓶中,投入上述粗品,加入17 g乙二醇单甲醚,25
±
5℃下搅拌2小时,抽滤,滤饼用适量乙二醇单甲醚淋洗,所得滤饼热风干燥(50
±
5℃),得到11.5 g棕色固体,即物质a1-5(分子量430.9,0.027mol),纯度98%,收率64.5%。
[0082]
(6)制备化合物a
[0083]
在烧瓶中加入10g物质a1-5(分子量430.9,0.023mol)、0.2l盐酸,冰浴下滴加10g亚硝酸钠的水溶液(含有亚硝酸钠3g,0.043mol),滴加完室温反应1h,将反应液缓慢倒入1l水中,析出黄色固体,搅拌1 h,过滤烘干,得4.0g化合物a(分子量395.4,0.01mol),纯度98%,收率43.1%。
[0084]
第三部分
[0085]
见图7-图10,本部分通过红外光谱、核磁共振氢谱(1h-nmr)和核磁共振碳谱(
13
c-nmr)、质谱对实施例1制备的化合物检测,以验证其结构。其中所述结构表征包括但不限于ir、uv、hrms、h-nmr、c-nmr。
[0086]
(1)红外光谱(ir)
[0087]
测试仪器型号:pe spectrum two;测试单位:上海张江药谷公共服务平台有限公
司;样品制备方法:atr法。
[0088]
结合图7和表1,红外光谱结果显示样品有明显的nh、s=o以及杂芳环,因此待标品的红外光谱数据与所述化合物的分子结构相符。
[0089]
表1化合物的红外光谱数据
[0090]
吸收峰(cm-1
)吸收强度振动类型基团3196.76w伸缩振动n-h3021.89w伸缩振动c-h1637.58~1385.90w伸缩振动苯杂环1169.39s不对称伸缩振动s=o1055s对称伸缩振动s=o
[0091]
(2)核磁共振氢谱(1h-nmr)和核磁共振碳谱(
13
c-nmr)
[0092]
氘代试剂:约9 mg实施例制备得到的化合物a溶解在0.50 ml dmso-d6中;可以将化合物的各基团序号标记为
[0093]
a)磁共振氢谱(1h-nmr)
[0094]
表2化合物的氢谱解析数据
[0095]
序号化学位移δh(ppm)multiplicity j[hz]质子数1h-1
h cosy12.93s3228.56s,br113////4////5////62.51s3/7////8////98.47d,3.21/10////119.61s,br1/12////137.99m117,1514////157.22-7.23m116,13,17167.22-7.23m115,17177.66-7.69m116,15,1318////
[0096]
b)磁共振氢谱核磁共振碳谱(
13
c-nmr)
[0097]
表3化合物的碳谱解析数据
[0098]
序号化学位移δc(ppm)碳原子数量
13
c-depthsqchmbc131.21ch31/2/////3170.21季c/14109.8,109.91季c/65151.81季c/6618.01ch36/7146.3,146.21季c/98148.6,146.11季c/99146.0,145.81ch9/10155.91季c/911/////12148.6or 139.81季c/1613116.31ch13/14148.6or 139.81季c/1615118.81ch15/16127.61ch16/17118.81ch17/18/////
[0099]
(3)质谱
[0100]
表4化合物的质谱数据
[0101]
质荷比(m/z)备注396.1[m+h]
+
[0102]
化合物的质谱结果显示,以上hrms(质谱联用法)分析的实测分子量为396.0591(m+1)与理论分子量396.0595(m+1)一致,由实测分子量推测分子式c
15h15
fn5o3s2与化合物的分子式c
15h14
fn5o3s2+h一致。化合物的质谱显示其分子量为395,综合红外光谱(ir),核磁共振氢谱(1h-nmr)、碳谱(
13
c-nmr)结果,可以确证样品分子结构与化合物的分子结构一致。
[0103]
综上所述,本发明的化合物及其制备工艺、应用以物质a1-5、盐酸、亚硝酸钠溶液为主原料进行第六反应制备所述化合物,可以用于药用化合物分析的标准品。首次提供了一种化合物及其合成方法,该化合物为ls007合成过程中的主要杂质之一,对ls007的质量控制具有重要意义。通过总计六次反应,以n-甲基硫脲、吡啶、3-氯-2,4-戊二酮为初始原料制备出所述化合物,可以作为系统适用性检测标准品。以上述依据本发明的理想实施例为启示,通过上述的说明内容,相关工作人员完全可以在不偏离本项发明技术思想的范围内,进行多样的变更以及修改。本项发明的技术性范围并不局限于说明书上的内容,必须要根据权利要求范围来确定其技术性范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1