一种异喹啉化合物的制备新方法与流程
1.本发明涉及药物合成技术领域,具体地来说,涉及一种异喹啉化合物的制备新方法。
背景技术:
2.罗沙司他(roxadustat)是由美国菲布罗根公司研制的全球首个小分子低氧诱导因子脯氨酰羟化酶抑制剂(hif
‑
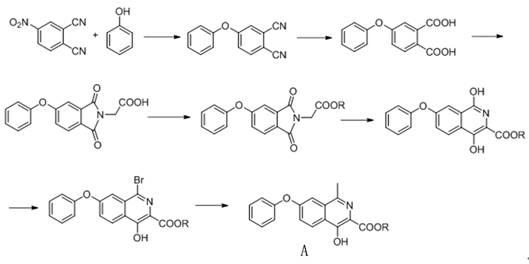
phi),其主要用于治疗正在接受透析治疗的患者因慢性肾病引起的贫血。因为该药还不具有标准的中文译名,故本技术人在此将其音译为“罗沙司他”。
3.罗沙司他具有异喹啉类化合物骨架结构,化学名为:2
‑
(4
‑
羟基
‑1‑
甲基
‑7‑
苯氧基
‑
异喹啉
‑3‑
甲酰胺基)乙酸;分子式:c
19
h
16
n2o5; cas号:808118
‑
40
‑
3;其化学结构式如下式a所示:,其中r为甲基、乙基、正丙基或异丙基。
4.而结构式b的异喹啉化合物,4
‑
羟基
‑1‑
甲基
‑7‑
苯氧基异喹啉
‑3‑
羧酸酯,是合成罗沙司他的关键中间体:,其中r为甲基、乙基、正丙基或异丙基。
5.现有技术公开了4
‑
羟基
‑1‑
甲基
‑7‑
苯氧基异喹啉
‑3‑
羧酸酯的多种制备方法,主要有:方法一:专利wo2004108681公开了以4
‑
硝基邻苯二甲腈为起始原料,经醚化、缩合、酯化、溴化和甲基化等多步反应得到结构式a的中间体4
‑ꢀ
羟基
‑1‑
甲基
‑7‑
苯氧基异喹啉
‑3‑
甲酸酯的技术路线,该技术路线的反应方程式如下所示:
,该方法需要大量的实验步骤来实现异喹啉环上1
‑
位甲基的引入,且引入过程需要贵金属进行催化,成本较高;同时,其在形成异喹啉环步骤的选择性差,生成异构体较多,需要柱层析提纯,收率低,成本高,不易规模化生产。
6.方法二:专利wo2014014834a公开了一种罗沙司他的新合成路线,该路线以5
‑
溴苯酞为起始原料,经过醚化、卤化和环化合成结构式a的中间体4
‑ꢀ
羟基
‑1‑
甲基
‑7‑
苯氧基异喹啉
‑3‑
甲酸酯,该合成路线的反应方程式如下所示:,该方法在第二步的开环反应过程中,需要使用大量的氯代试剂(12
‑
15eq),且反应产生大量酸气,环境污染较大,对设备要求高,同时其废液处理又需使用大量碱液进行中和,环保压力大,并且后续仍需三步反应来实现异喹啉环上1
‑
位甲基的引入,总路线长,工序繁杂,耗时较多,不适合大规模生产。
7.方法三:专利cn104892509a公布了一种以酪氨酸为起始原料,经酯化、醚化、环化以及羟基化得到结构式a的中间体4
‑ꢀ
羟基
‑1‑
甲基
‑7‑
苯氧基异喹啉
‑3‑
甲酸酯的合成路线,该合成路线的反应方程式如下所示:
,该方法的第二步醚化反应所生成的伯胺很不稳定,在空气中易被氧化;同时,第三步环化反应收率较低;另外,第四步引入酚羟基的过程中,容易生成氨基取代的副产物,给最终产物的纯化带来较大的困难。
8.方法四:cn106478504a专利中公开了以3
‑
溴苯乙酮为起始原料,经过醚化、还原胺化、缩合以及环化反应合成结构式a的中间体4
‑ꢀ
羟基
‑1‑
甲基
‑7‑
苯氧基异喹啉
‑3‑
甲酸酯的技术路线,该技术路线的反应方程式如下所示:,该方法的反应步骤较短,整体相对简单,但是第三步需用到酮基丙二酸二甲酯,该试剂价格昂贵,不易获得;第四步环化温度很高,接近200℃,工业化操作难度较大。
9.方法五:专利cn106478503a公开结构式a的中间体的制备方法,其制备方法的反应方程式如下所示:,该方法中先引入甲基,再构建异喹啉环,解决了异喹啉环引入甲基的难点,但仍存在明显不足:第一步反应用到了高能量的肼试剂,不利于安全操作;第二步反应用到了昂贵的氧化试
剂碘苯二乙酸,且氧化反应也不利于规模化的安全生产。
10.此外,专利cn112375057a采用通过4
‑
溴邻苯二甲酸酐(化合物i)与甲基格氏试剂加成得到中间体ii,中间体ii先经过硼氢化钠还原,再经酸性条件下环合生成中间体iii,中间体iii和苯酚发生芳基烷基化反应得到罗沙司他中间体iv[3
‑
甲基
‑5‑
苯基异苯并呋喃
‑
1(3h)
‑
酮]的技术路线。该现有技术中虽然以4
‑
溴邻苯二甲酸酐为原料,且同样提前引入了甲基,但是解决不了制备得到4
‑
羟基
‑1‑
甲基
‑7‑
苯氧基异喹啉
‑3‑
羧酸酯的技术问题。
技术实现要素:
[0011]
本发明的目的在于提供一种异喹啉化合物即4
‑
羟基
‑1‑
甲基
‑7‑
苯氧基异喹啉
‑3‑
羧酸酯的制备新方法,以解决上述背景技术中提出的现有方法的异喹啉环上1
‑
甲基引入较难,氯代试剂等试剂的用量多,生产成本高,反应路线繁琐,操作安全性低,各中间体纯化不方便同时反应后处理困难,环境污染大,反应条件要求高,生产效率低,不适合规模化生产且整体收率一般的技术问题。
[0012]
本发明采取以下技术方案:一种异喹啉化合物的制备新方法,具体操作步骤如下:1)中间体int1的制备:以邻苯二甲酸酐为起始原料,溶于碱性溶剂中,添加烷基化试剂,开环并与摩尔比1.2~1.5倍的烷基化试剂进行反应,反应结束后,加入盐酸酸化,制备得到中间体int1,所述碱性溶剂选自乙二胺、丙二胺或三乙胺,所述烷基化试剂选自丙二酸、丙二酸二甲酯或丙二酸二乙酯;2)中间体int2的制备:中间体int1在碱的作用下,经过摩尔比1.1~1.5倍的三乙酰氧基硼氢化钠还原,再经酸性条件下环合生成中间体int2,所述碱选自氢氧化钠、氢氧化锂或氢氧化钾;3)中间体int3的制备:中间体int2溶于溶剂中,在
‑
20~
‑
15℃下,与摩尔比1.1~1.5倍的溴代试剂反应生成中间体int3,所述溴代试剂选自1,3
‑
二溴
‑
5,5
‑
二甲基海因、n
‑
溴代琥珀酰亚胺或溴素,所述溶剂选自n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺或n
‑
甲基吡咯烷酮、二氯甲烷,;4)中间体int4的制备:中间体int3溶于溶剂中,在摩尔比0.1~1倍的相转移催化剂、摩尔比0.1~1倍的酸催化剂和摩尔比1.1~2.0倍的氯代试剂存在下,开环生成酰氯,酰氯与醇接触生成中间体int4,所述溶剂选自甲苯、二甲苯或氯苯,所述相转移催化剂选自苄基三乙基氯化铵、四丁基溴化铵或四丁基硫酸氢铵,所述酸催化剂选自三氟化硼乙醚、硼酸三甲酯或硼酸,所述氯代试剂为氯化亚砜,所述醇选自甲醇、乙醇、正丙醇或异丙醇;5)中间体int5的制备:中间体int4溶于溶剂中,在摩尔比2.5~3.0倍的碱剂ⅰ及摩尔比0.2~0.8倍的碘化物的催化下,与摩尔比1.1~2.0倍的对甲苯磺酰甘氨酸甲酯反应生成中间体int 4',反应完成后抽滤,向滤液中加入摩尔比0.9~1.8倍的碱剂ⅱ后进行缩合,并脱保护得到中间体int5,所述溶剂选自n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺或n
‑
甲基吡咯烷酮,所述碱剂ⅰ选自碳酸钠、碳酸钾或碳酸铯,所述碘化物选自碘化钠或碘化钾,所述碱剂ⅱ选自甲醇钠或乙醇钠;6)异喹啉化合物b的制备:中间体int5在摩尔比3.0~4.0倍的缚酸剂、摩尔比0.2~0.5倍的催化剂存在下,与摩尔比1.1~1.5倍的苯酚在溶剂中发生偶联反应生成异喹啉化合物b,即4
‑
羟基
‑1‑
甲基
‑7‑
苯氧基异喹啉
‑3‑
羧酸酯,所述溶剂选自n,n
‑
二甲基甲酰胺、
n,n
‑
二甲基乙酰胺或n
‑
甲基吡咯烷酮,所述缚酸剂选自碳酸钠、碳酸钾或碳酸铯,所述催化剂选自溴化亚铜或碘化亚铜;所述制备新方法的反应方程式如下:,其中,式b中的r为甲基、乙基、正丙基或异丙基。
[0013]
与现有技术相比,本发明的有益效果为:1、本发明以邻苯二甲酸酐为起始原料,经开环烷基化制得中间体int1,int1经还原环合得到int2,int2经溴代得到int3,int3经开环酯化得到int4,int4经环合得到int5,int5与苯酚偶联得到异喹啉化合物即4
‑
羟基
‑1‑
甲基
‑7‑
苯氧基异喹啉
‑3‑
羧酸酯,原料价格低廉易得,反应路线简单,操作安全度高,后处理方便,各中间体纯化方便,整体收率较高,适合规模化生产;2、本发明中的步骤1)、2)提前构建了甲基,解决了后续异喹啉环上1
‑
甲基引入较难的问题,且反应条件温和,避免了氧化试剂碘苯二乙酸的使用,更适合工业规模化生产;3、本发明中,步骤4)的开环酯化反应在步骤6)的偶联反应之前进行,大幅度地降低了氯代试剂的用量,解决了使用大量的氯代试剂(12
‑
15eq)的难题,减小了环境污染,降低了固废产出;4. 本发明中的步骤5)中,中间体int4’仅仅经简单的抽滤处理后便继续反应,两步并做一步反应,极大地缩短了工序,节约了生产时间,提高了生产效率。
具体实施方式
[0014]
实施例12
‑
乙酰基苯甲酸(int1)的制备:,
向2l反应瓶中,依次加入邻苯二甲酸酐(230g,1.55mol)、丙二酸(32.2g,0.31mol)和三乙胺(320ml),搅拌下升温至78
‑
82℃,到达温度后分批加入丙二酸(5*32.2g,1.55mol),加完保温78
‑
82反应10h。反应液降温至25
‑
30℃,控温滴加4n hcl(1.22l),滴加过程中大量固体析出,滴完后搅拌0.5h,过滤,滤饼用水洗涤,真空烘箱50℃干燥得到int1(233g,91.7%)。
[0015]
实施例22
‑
乙酰基苯甲酸(int1)的制备:,向10l反应瓶中,依次加入邻苯二甲酸酐(1.2kg,8.1mol)、丙二酸(210.7g,2.025mol)和三乙胺(1.6l),搅拌下升温至78
‑
82℃,到达温度后分批加入丙二酸(5*210.7g,10.125mol),加完保温78
‑
82反应24h。反应液降温至25
‑
30℃,控温滴加4n hcl(6.1l),滴加过程中大量固体析出,滴完后搅拌0.5h后,过滤,滤饼用水洗涤,真空烘箱50℃干燥得到int1(2250g,88.6%)。
[0016]
实施例32
‑
乙酰基苯甲酸(int1)的制备:,向10l反应瓶中,依次加入邻苯二甲酸酐(1.2kg,8.1mol)、丙二酸(175.8g,1.69mol)和三乙胺(1.6l),搅拌下升温至78
‑
82℃,到达温度后分批加入丙二酸(5*175.8g,8.45mol),加完保温78
‑
82反应24h。反应液降温至25
‑
30℃,控温滴加4n hcl(6.1l),滴加过程中大量固体析出,滴完后搅拌0.5h后,过滤,滤饼用水洗涤,真空烘箱50℃干燥得到int1(2293g,90.3%)。
[0017]
实施例43
‑
甲基苯酞(int2)的制备:,向1l反应瓶中,加入5%氢氧化钠溶液(320g)和2
‑
乙酰基苯甲酸(32.8g,0.2mol),常温搅拌15min,逐渐降温至0
‑
10℃,分批加入三乙酰氧基硼氢化钠(46.6g,0.22mol),加完保温反应5h;反应完成后加入二氯甲烷,用6n hcl调节ph=2,搅拌充分后静置分层,分液,水相用二氯甲烷提取两次,合并有机相,用饱和碳酸氢钠溶液洗涤至中性,纯化水洗涤一次,
无水硫酸镁干燥,减压蒸馏,得油状物,用mtbe回流打浆,过滤,真空烘箱50℃干燥得到int2(26.2g,88.5%)。
[0018]
实施例53
‑
甲基苯酞(int2)的制备:,向10l反应瓶中,加入5%氢氧化钠溶液(3.2kg)和2
‑
乙酰基苯甲酸(328g,2.0mol),常温搅拌1h,逐渐降温至0
‑
10℃,分批加入三乙酰氧基硼氢化钠(551g,2.6mol),加完保温反应8h;反应完成后加入二氯甲烷,用6n hcl调节ph=2,搅拌充分后静置分层,分液,水相用二氯甲烷提取两次,合并有机相,用饱和碳酸氢钠溶液洗涤至中性,纯化水洗涤一次,减压蒸馏,得油状物,用mtbe回流打浆,过滤,真空烘箱50℃干燥得到int2(254.8g,86.0%)。
[0019]
实施例63
‑
甲基苯酞(int2)的制备:,向10l反应瓶中,加入5%氢氧化钠溶液(3.2kg)和2
‑
乙酰基苯甲酸(328g,2.0mol),常温搅拌1h,逐渐降温至0
‑
10℃,分批加入三乙酰氧基硼氢化钠(635.8g,3.0mol),加完保温反应8h;反应完成后加入二氯甲烷,用6n hcl调节ph=2,搅拌充分后静置分层,分液,水相用二氯甲烷提取两次,合并有机相,用饱和碳酸氢钠溶液洗涤至中性,纯化水洗涤一次,减压蒸馏,得油状物,用mtbe回流打浆,过滤,真空烘箱50℃干燥得到int2(264.8g,89.4%)。
[0020]
实施例75
‑
溴
‑3‑
甲基
‑
苯酞(int3)的制备: ,向1l反应瓶中加入3
‑
甲基苯酞(44.4g,0.3mol)和n,n
‑
二甲基甲酰胺(450ml),降温至
‑
20~
‑
15℃,一次性加入n
‑
溴代琥珀酰亚胺(58.7g,0.33mol),保温反应3h,反应结束后加入1n氢氧化钠溶液,逐渐升温至常温搅拌1h,固体逐渐析出,继续搅拌析晶2h,过滤,滤饼用少量水洗涤,真空烘箱50℃干燥得到int3(52.3g,76.8%)。
[0021]
实施例85
‑
溴
‑3‑
甲基
‑
苯酞(int3)的制备:
ꢀ
,向10l反应瓶中加入3
‑
甲基苯酞(444g,4.0mol)和n,n
‑
二甲基甲酰胺(4.5l),降温至
‑
20~
‑
15℃,一次性加入n
‑
溴代琥珀酰亚胺(1068g,6.0mol),保温反应6h,反应结束后加入1n氢氧化钠溶液,逐渐升温至常温搅拌1h,固体逐渐析出,继续搅拌析晶2h,过滤,滤饼用少量水洗涤,真空烘箱50℃干燥得到int3(536.6g,78.8%)。
[0022]
实施例95
‑
溴
‑3‑
甲基
‑
苯酞(int3)的制备: ,向10l反应瓶中加入3
‑
甲基苯酞(444g,4.0mol)和n,n
‑
二甲基甲酰胺(4.5l),降温至
‑
20~
‑
15℃,一次性加入n
‑
溴代琥珀酰亚胺(925.5g,5.2mol),保温反应6h,反应结束后加入1n氢氧化钠溶液,逐渐升温至常温搅拌1h,固体逐渐析出,继续搅拌析晶2h,过滤,滤饼用少量水洗涤,真空烘箱50℃干燥得到int3(528.4g,77.6%)。
[0023]
实施例104
‑
溴
‑2‑
(1
‑
氯乙基)苯甲酸甲酯(int4)的制备: ,向1l反应瓶中依次投入5
‑
溴
‑3‑
甲基
‑
苯酞(53g,0.23mol)、苄基三乙基氯化铵(5.3g,0.023mol)和甲苯(530ml),搅拌下一次性加入氯化亚砜(30.1g,0.253mol)、三氟化硼乙醚(3.26g,0.023 mol),升温至90
‑
100℃回流反应16h。反应完毕降温至室温,减压蒸馏除去氯化亚砜和甲苯,向残留物中加入甲醇,再升温至50~55℃反应1h,减压蒸出溶剂,加入乙酸乙酯和5%氢氧化钠溶液,萃取分层,有机相用5%氢氧化钠溶液洗涤两次,合并有机相干燥蒸干后得棕红色油状物int4(63.5g,99.5%)。
[0024]
实施例114
‑
溴
‑2‑
(1
‑
氯乙基)苯甲酸甲酯(int4)的制备:,
向3l反应瓶中依次投入5
‑
溴
‑3‑
甲基
‑
苯酞(159g,0.7mol)、苄基三乙基氯化铵(159g,0.7mol)和甲苯(1.6l),搅拌下一次性加入氯化亚砜(166.6g,1.4 mol)、三氟化硼乙醚(99.3g,0.7 mol),升温至90
‑
100℃回流反应16h。反应完毕降温至室温,减压蒸馏除去氯化亚砜和甲苯,向残留物中加入甲醇,再升温至50~55℃反应1h,减压蒸出溶剂,加入乙酸乙酯和5%氢氧化钠溶液,萃取分层,有机相用5%氢氧化钠溶液洗涤两次,合并有机相干燥蒸干后得棕红色油状物int4(190g,97.8%)。
[0025]
实施例124
‑
溴
‑2‑
(1
‑
氯乙基)苯甲酸甲酯(int4)的制备:,向3l反应瓶中依次投入5
‑
溴
‑3‑
甲基
‑
苯酞(159g,0.7mol)、苄基三乙基氯化铵(87.7g,0.385mol)和甲苯(1.6l),搅拌下一次性加入氯化亚砜(129g,1.085 mol)、三氟化硼乙醚(54.6g,0.385 mol),升温至90
‑
100℃回流反应16h。反应完毕降温至室温,减压蒸馏除去氯化亚砜和甲苯,向残留物中加入甲醇,再升温至50~55℃反应1h,减压蒸出溶剂,加入乙酸乙酯和5%氢氧化钠溶液,萃取分层,有机相用5%氢氧化钠溶液洗涤两次,合并有机相干燥蒸干后得棕红色油状物int4(191.3g,98.5%)。
[0026]
实施例134
‑
羟基
‑1‑
甲基
‑7‑
溴
‑
异喹啉
‑3‑
羧酸甲酯(int5)的制备:,向1l反应瓶中加入dmf(500ml),依次投入4
‑
溴
‑2‑
(1
‑
氯乙基)苯甲酸甲酯(83.2g,0.3mol)、对苯甲磺酰基甘氨酸甲酯(80.3g,0.33mol)、碳酸钾(103.5g,0.75mol)和碘化钠(9.0g,0.06mol),升温至55~60℃反应1h,反应完直接抽滤,滤饼用少量dmf洗涤,滤液转移至另一1l反应瓶中,降温至10~15℃,滴加24%甲醇钠/甲醇溶液(60g,0.27mol),滴完保温反应1h,将反应液倒入水中,搅拌下控温10~20℃滴加冰醋酸调节ph=7,呈乳白色悬浊液,继续保温析晶1h后抽滤,滤饼用水洗涤,真空烘箱50℃干燥得到int5(78g,87.8%)。
[0027]
实施例144
‑
羟基
‑1‑
甲基
‑7‑
溴
‑
异喹啉
‑3‑
羧酸甲酯(int5)的制备:,
向10l反应瓶中加入dmf(5l),依次投入4
‑
溴
‑2‑
(1
‑
氯乙基)苯甲酸甲酯(832g,3mol)、对苯甲磺酰基甘氨酸甲酯(948g,3.9mol)、碳酸钾(1.14kg,8.25mol)和碘化钠(225g,1.5mol),升温至55~60℃反应2h,反应完直接抽滤,滤饼用少量dmf洗涤,滤液转移至另一10l反应瓶中,降温至10~15℃,滴加甲醇钠/甲醇溶液,滴完保温反应2h,将反应液倒入水中,搅拌下控温10~20℃滴加冰醋酸调节ph=7,呈乳白色悬浊液,继续保温析晶2h后抽滤,滤饼用水洗涤,真空烘箱50℃干燥得到int5(795g,89.6%)。
[0028]
实施例154
‑
羟基
‑1‑
甲基
‑7‑
溴
‑
异喹啉
‑3‑
羧酸甲酯(int5)的制备:,向10l反应瓶中加入dmf(5l),依次投入4
‑
溴
‑2‑
(1
‑
氯乙基)苯甲酸甲酯(832g,3mol)、对苯甲磺酰基甘氨酸甲酯(1460g,6mol)、碳酸钾(1.24kg,9mol)和碘化钠(360g,2.4mol),升温至55~60℃反应2h,反应完直接抽滤,滤饼用少量dmf洗涤,滤液转移至另一10l反应瓶中,降温至10~15℃,滴加24%甲醇钠/甲醇溶液(911g,4.05mol),滴完保温反应2h,将反应液倒入水中,搅拌下控温10~20℃滴加冰醋酸调节ph=7,呈乳白色悬浊液,继续保温析晶2h后抽滤,滤饼用水洗涤,真空烘箱50℃干燥得到int5(771g,86.8%)。
[0029]
实施例164
‑
羟基
‑1‑
甲基
‑7‑
苯氧基异喹啉
‑3‑
羧酸甲酯(b)的制备:,向1l反应瓶中投入dmf(500ml),再依次投入4
‑
羟基
‑1‑
甲基
‑7‑
溴
‑
异喹啉
‑3‑
羧酸甲酯(59.2g,0.2mol)、苯酚(20.7g,0.22mol)、溴化亚铜(5.7g,0.04mol)、和碳酸钾(82.8g,0.6mol),氮气置换三次,升温至90~100℃反应16h,反应完毕向反应液中加入水,搅拌1h后过滤,滤饼用乙酸乙酯回流溶解经活性炭脱色后抽滤,滤液蒸干后得粗品,粗品经四氢呋喃精制得到b(45.5g,73.6%)。
[0030]
实施例174
‑
羟基
‑1‑
甲基
‑7‑
苯氧基异喹啉
‑3‑
羧酸甲酯(b)的制备:,
向10l反应瓶中投入dmf(5l),再依次投入4
‑
羟基
‑1‑
甲基
‑7‑
溴
‑
异喹啉
‑3‑
羧酸甲酯(592g,2mol)、苯酚(244.7g,2.6mol)、溴化亚铜(100.4g,0.7mol)、和碳酸钾(966g,7.0mol),氮气置换三次,升温至90~100℃反应16h,反应完毕向反应液中加入水,搅拌1h后过滤,滤饼用乙酸乙酯回流溶解经活性炭脱色后抽滤,滤液蒸干后得粗品,粗品经四氢呋喃精制得到b(439.2g,71%)。
[0031]
实施例184
‑
羟基
‑1‑
甲基
‑7‑
苯氧基异喹啉
‑3‑
羧酸甲酯(b)的制备:,向10l反应瓶中投入dmf(5l),再依次投入4
‑
羟基
‑1‑
甲基
‑7‑
溴
‑
异喹啉
‑3‑
羧酸甲酯(592g,2mol)、苯酚(282.3g,3.0mol)、溴化亚铜(143.5g,1.0mol)、和碳酸钾(1104g,8.0mol),氮气置换三次,升温至90~100℃反应16h,反应完毕向反应液中加入水,搅拌1h后过滤,滤饼用乙酸乙酯回流溶解经活性炭脱色后抽滤,滤液蒸干后得粗品,粗品经四氢呋喃精制得到b(429.3g,69.4%)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1