氧化半日花烷型二萜类化合物及其分离方法和应用
1.本发明涉及天然的植物提取物,具体涉及氧化半日花烷型二萜类化合物及其分离方法和应用。
背景技术:
2.裸花紫珠(callicarpa nudiflora hook.et arn.)又名赶风柴、节节红,是马鞭草科(verbenacase)紫珠属(callicarpa)植物,为海南省道地药材,也是黎族地区的经典药材。裸花紫珠地上部位均可入药,有消炎解毒,散瘀消肿,抗菌止血等功效,主治化脓性炎症、创伤性出血、消化道和呼吸道感染、烧烫伤等症。临床辅助用于皮肤科、妇科、五官科、外科等各类术后出血型疾病。现已被2015年版《中国药典》新增中药品种收录。
3.裸花紫珠的化学成分研究报道主要为黄酮(苷)类、苯丙素(苷)类、二萜、三萜、环烯醚萜类、酚酸类等多种类的化合物,且都具有较好的生物活性。wang z.h.等报道的二萜类化合物(wang z.h.,xu h.j.,zhai y.y.,et al.three new labdane
‑
type diterpenoids from callicarpa macrophylla vahl.,natural produvt research,doi.org/10.1080/14786419.2018.1509336)”,部分半日花烷型二萜类化合物具有抗炎作用,但效果欠佳。裸花紫珠中含有大量具有生物活性的化合物,本发明旨在对裸花紫珠化学成分进一步分离研究,以期得到新结构的具有显著抗炎作用的半日花烷型二萜类化合物,充分发挥裸花紫珠的药用价值。
技术实现要素:
4.鉴于现有技术的不足,本发明提供一种新结构的氧化半日花烷型二萜类化合物,具有显著的抗炎作用,并提供目标化合物的分离方法。
5.本发明技术方案如下:
6.本发明提供氧化半日花烷型二萜类化合物,其结构分别为:
[0007][0008]
本发明还提供了化合物i和ii的分离方法,包括以下步骤:
[0009]
(1)将干燥裸花紫珠叶片粉碎,将裸花紫珠粉末使用水煮沸,保持沸腾提取得到提取液,减压浓缩得粗提物;
[0010]
(2)将步骤(1)的粗提物用水稀释制成悬浮液后,依次用二氯甲烷、乙酸乙酯萃取,合并有机相,减压浓缩得浸膏;
[0011]
(3)取步骤(2)乙酸乙酯萃取得到的浸膏,先经硅胶柱层析,采用氯仿和丙酮混合溶剂作为洗脱剂进行梯度洗脱,洗脱梯度为体积比(100:1)~(1:10),根据极性大小共得到8个组分,即fr.1~fr.8;
[0012]
(4)将fr.2先经正相硅胶柱层析,采用石油醚和乙酸乙酯混合溶剂作为洗脱剂进行梯度洗脱,洗脱梯度为体积比(10:1)~(1:10),减压浓缩后再经ods反相柱分离,采用甲醇和水混合溶剂作为洗脱剂进行梯度洗脱,洗脱梯度为体积比(10:90)
‑
(90:10),根据极性大小共得到7个组分,即fr.2a~fr.2f;取fr.2d再经sephadex lh
‑
20凝胶柱层析,洗脱剂为甲醇,减压浓缩后再经高效液相色谱hplc制备,依次得到化合物ⅰ和化合物ⅱ。
[0013]
优选的,步骤(1)中,所述提取次数为2次以上,每次提取1~3h,合并提取液;步骤(2)中,依次用二氯甲烷、乙酸乙酯各萃取3次以上。
[0014]
优选的,在步骤(1)中,所述水为纯净水,其用量为每千克裸花紫珠粉末用2~3l。
[0015]
优选的,在步骤(1)中,加热至水沸腾,保持沸腾提取1
‑
2h,所述提取次数为2次以上。
[0016]
优选的,在步骤(2)中,依次用二氯甲烷、乙酸乙酯各萃取3次以上,合并萃取液。
[0017]
优选的,在步骤(2)中,水的用量为每100克粗提物加水300~400ml,每次萃取的有机溶剂的体积用量是水体积的1.2~1.3倍。
[0018]
优选的,在步骤(3)中,氯仿
‑
丙酮混合溶剂的洗脱梯度为100:1、80:1、50:1、20:1、10:1、5:1、1:1、1:10,每个梯度收集3个柱体积,每个梯度得到一个组分,共得到8个组分,即fr.1~fr.8。
[0019]
优选的,在步骤(4)中,石油醚
‑
乙酸乙酯混合溶剂的洗脱梯度为10:1、5:1、1:1、1:10,每个梯度洗脱2~5个柱体积。
[0020]
优选的,在步骤(4)中,ods反相洗脱比例为甲醇:水(v:v)10:90、30:70、50:50、60:40、70:30、80:20、90:10,每个梯度洗脱3~5个柱体积。
[0021]
优选的,在步骤(4)中,sephadex lh
‑
20凝胶柱层析,洗脱剂为甲醇,洗脱3~6个柱体积。
[0022]
优选的,在步骤(4)中,高效液相色谱的条件为:色谱柱waters c
18
,流速为2ml/min,流动相为体积比60:40的乙腈:水。
[0023]
优选的,所述裸花紫珠为采集于海南五指山的裸花紫珠。
[0024]
本发明分离得到的半日花烷型二萜类化合物在制备预防或治疗炎症的药物中的应用。优选的,所述炎症为系统性炎症反应综合征、支气管炎、肺炎、胃炎或肠炎。
[0025]
与现有技术相比,本发明的有益效果是:
[0026]
本发明从裸花紫珠叶子中提取分离出氧化半日花烷型二萜类化合物i和ii,对比同类型化合物,其具有较好的抗炎效果。具体地,本发明通过水提、蒸馏水溶解分散、极性溶剂萃取、溶剂梯度洗脱和液相色谱分离等多级分离提取方法,从裸花紫珠浸膏中得到新氧化半日花烷型二萜类化合物。
附图说明
[0027]
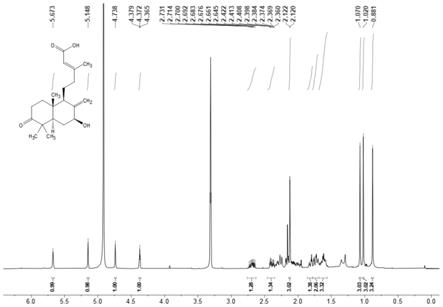
图1:化合物i的1h
‑
nmr谱(meod
‑
d4)
[0028]
图2:化合物i的
13
c
‑
nmr谱(meod
‑
d4)
[0029]
图3:化合物i的dept(135
°
)谱(meod
‑
d4)
[0030]
图4:化合物i的1h
‑1h cosy谱(meod
‑
d4)
[0031]
图5:化合物i的hsqc谱(meod
‑
d4)
[0032]
图6:化合物i的hmbc谱(meod
‑
d4)
[0033]
图7:化合物i的noesy谱(meod
‑
d4)
[0034]
图8:化合物i的hresims谱
[0035]
图9:化合物ii的1h
‑
nmr谱(meod
‑
d4)
[0036]
图10:化合物ii的
13
c
‑
nmr谱(meod
‑
d4)
[0037]
图11:化合物ii的dept(135
°
)谱(meod
‑
d4)
[0038]
图12:化合物ii的1h
‑1h cosy谱(meod
‑
d4)
[0039]
图13:化合物ii的hsqc谱(meod
‑
d4)
[0040]
图14:化合物ii的hmbc谱(meod
‑
d4)
[0041]
图15:化合物ii的hmbc局部放大谱(meod
‑
d4)
[0042]
图16:化合物ii的noesy谱(meod
‑
d4)
[0043]
图17:化合物ii的hresims谱
具体实施方式
[0044]
为了更好理解本发明技术内容,下面提供具体实施例,对本发明做进一步的说明。
[0045]
本发明实施例所用的实验方法如无特殊说明,均为常规方法。
[0046]
本发明实施例所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0047]
本发明实验材料裸花紫珠采集于海南五指山,所用部位为叶子。
[0048]
实施例1高度修饰半日花烷型二萜类化合物的制备
[0049]
包括以下步骤:
[0050]
(1)将干燥裸花紫珠叶片粉碎,按每千克裸花紫珠用2l水加热至沸腾,保持沸腾提取2次,每次提取1.5h,合并提取液,减压浓缩得粗提物(约300g);
[0051]
(2)取每100克粗提物加蒸馏水300ml稀释制成悬浮液后,依次用二氯甲烷、乙酸乙酯各萃取3次,合并有机相,减压浓缩得浸膏;每次萃取的有机溶剂的体积用量是水体积的1.2倍。
[0052]
(3)取步骤(2)乙酸乙酯萃取得到的浸膏(约10g),先经硅胶柱层析,采用氯仿
‑
丙酮混合溶剂进行梯度洗脱,洗脱梯度为体积比100:1、80:1、50:1、20:1、10:1、5:1、1:1、1:10,每个梯度收集3个柱体积,每个梯度得到一个组分,共得到8个组分,即fr.1~fr.8。
[0053]
(4)将fr.2先经正相硅胶柱层析,采用石油醚
‑
乙酸乙酯混合溶剂作为洗脱剂进行梯度洗脱,洗脱梯度为体积比10:1、5:1、1:1、1:10,每个梯度洗脱3个柱体积;再经ods反相分离,洗脱比例为meoh:h2o(v:v)10:90、30:70、50:50、60:40、70:30、80:20、90:10,每个梯度洗脱4个柱体积,根据极性大小共得到7个组分,即fr.2a~fr.2g;取fr.2d再经sephadex lh
‑
20凝胶柱层析,洗脱剂为meoh,洗脱3个柱体积;减压浓缩后再经高效液相色谱hplc制备,得到化合物i和ii;高效液相色谱的条件为:色谱柱waters c
18
,流速为2ml/min,流动相为体积比60:40的mecn:h2o。
[0054]
实施例2高度修饰半日花烷型二萜类化合物的制备
[0055]
包括以下步骤:
[0056]
(1)将干燥裸花紫珠片粉碎,按每千克裸花紫珠粉末用2.5l水加热至沸腾,保持沸腾提取3次,每次提取1h,合并提取液,减压浓缩得粗提物(约320g);
[0057]
(2)将每100克粗提物加水400ml稀释制成悬浮液后,依次用二氯甲烷、乙酸乙酯各萃取4次,合并有机相,减压浓缩得浸膏;每次萃取的有机溶剂的体积用量是水体积的1.3倍。
[0058]
(3)取步骤(2)乙酸乙酯萃取得到的浸膏(约12g),先经硅胶柱层析,采用氯仿
‑
丙酮混合溶剂进行梯度洗脱,洗脱梯度为体积比100:1、80:1、50:1、20:1、10:1、5:1、1:1、1:10,每个梯度收集3个柱体积,每个梯度得到一个组分,共得到8个组分,即fr.1~fr.8。
[0059]
(4)将fr.2先经正相硅胶柱层析,采用石油醚
‑
乙酸乙酯混合溶剂作为洗脱剂进行梯度洗脱,洗脱梯度为体积比10:1、5:1、1:1、1:10,每个梯度洗脱4个柱体积,再经ods反相分离,洗脱比例为meoh:h2o(v:v)10:90、30:70、50:50、60:40、70:30、80:20、90:10,每个梯度洗脱4个柱体积,根据极性大小共得到7个组分,即fr.2a~fr.2g;取fr.2d再经sephadex lh
‑
20凝胶柱层析,洗脱剂为meoh,洗脱6个柱体积,减压浓缩后再经高效液相色谱hplc制备,得到化合物i和ii;高效液相色谱的条件为:色谱柱waters c
18
,流速为2ml/min,流动相为体积比60:40的mecn:h2o。
[0060]
实施例3高度修饰半日花烷型二萜类化合物的结构鉴定
[0061]
运用波谱(1h nmr,
13
c nmr,hsqc,hmbc,noesy)和ms等现代结构鉴定技术,确定实施例1和实施例2所得化合物i和ii的化学结构。
[0062]
结构鉴定数据如下:
[0063]
化合物i:为无色油状物,易溶于甲醇。经高分辨质谱hresi(
‑
)ms(m/z 357.2032[m+na]
+
,理论值357.2036)确定其分子式为c
20
h
30
o4;根据1h,
13
c及二维核磁共振数据确定其结构,骨架类型为半日花烷型二萜。经甲醇缓慢挥发,得到了该化合物的单晶,如下图所示,确定绝对构型为5r,7s,9r,10r,即该化合物为
[0064]3‑
oxo
‑7‑
hydroxy
‑
8(17),13
‑
ent
‑
labdadien
‑
15
‑
oic acid,将其命名为callnudoid a,其1h和
13
c nmr数据归属见表1。[400mhz(1h),100mhz(
13
c),溶剂:meod
‑
d4]。
[0065][0066]
化合物i的1h
‑
nmr谱(meod
‑
d4)如图1所示,
[0067]
化合物i的
13
c
‑
nmr谱(meod
‑
d4)如图2所示,
[0068]
化合物i的dept(135
°
)谱(meod
‑
d4)如图3所示,
[0069]
化合物i的1h
‑1h cosy谱(meod
‑
d4)如图4所示,
[0070]
化合物i的hsqc谱(meod
‑
d4)如图5所示,
[0071]
化合物i的hmbc谱(meod
‑
d4)如图6所示,
[0072]
化合物i的noesy谱(meod
‑
d4)如图7所示,
[0073]
化合物i的hresims谱如图8所示。
[0074]
化合物ii:为无色晶状物,易溶于甲醇。经高分辨质谱hresi(
‑
)ms(m/z 359.2193[m+na]
+
,理论值359.2198)确定其分子式为c
15
h
24
o3;根据1h,
13
c及二维核磁共振数据确定其结构,骨架类型为高度修饰半日花烷型二萜,3
‑
oxo
‑8‑
hydroxy
‑
13
‑
ent
‑
labdadien
‑
15
‑
oic acid,将其命名为callnudoid b。其1h和
13
c nmr数据归属见表1。[400mhz(1h),100mhz(
13
c),溶剂:meod
‑
d4]。
[0075]
化合物ii的1h
‑
nmr谱(meod
‑
d4)如图9所示,
[0076]
化合物ii的
13
c
‑
nmr谱(meod
‑
d4)如图10所示,
[0077]
化合物ii的dept(135
°
)谱(meod
‑
d4)如图11所示,
[0078]
化合物ii的1h
‑1h cosy谱(meod
‑
d4)如图12所示,
[0079]
化合物ii的hsqc谱(meod
‑
d4)如图13所示,
[0080]
化合物ii的hmbc谱(meod
‑
d4)如图14所示,
[0081]
化合物ii的hmbc局部放大谱(meod
‑
d4)如图15所示,
[0082]
化合物ii的noesy谱(meod
‑
d4)如图16所示,
[0083]
化合物ii的hresims谱如图17所示。
[0084]
表1.化合物i和ii的1h
‑
nmr和
13
c
‑
nmr(400,100mhz)数据
[0085]
[0086][0087]
经以上分析,确定化合物i和ii结构为:
[0088][0089]
实施例4药理活性实验
[0090]
实验材料:
[0091]
细胞:小鼠单核巨噬细胞raw264.7。
[0092]
细胞培养液:含10%胎牛血清(fbs)的dmem培养基,脂多糖(lps)碳水化合物。
[0093]
no检测试剂盒:普利莱(applygen),货号:e1030。
[0094]
实验方法:
[0095]
诱导:raw264.7细胞用含10%fbs的dmem培养液于37℃、5%co2培养箱中常规培养。细胞按1
×
105/ml、200μl/孔接种于96孔板中,分别设置空白对照组、lps诱导组、受试药物高、中、低(50,25,12.5μm)剂量组,以吲哚美辛(indomethacin)为阳性对照组,置于37℃、5%co2细胞培养箱中贴壁24 h后。
[0096]
检测:吸取50μl上清作为待测液于96孔板中,根据试剂盒说明书的检测方法,依次加入50μl试剂a,50μl试剂b,采用酶标仪在540 nm处测od值。
[0097]
化合物的抗炎活性结果如表2所示,从结果可知,化合物i和ii都表现出明显的抗炎效果,ic
50
分别为10.8
±
0.34μm和12.7
±
0.42μm。
[0098]
表2化合物i和ii对raw264.7细胞抗炎活性(n=3).
[0099][0100]
以上内容是结合具体的实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。对于本发明所属技术领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演或替换。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1