一种炎症小体核苷酸结合寡聚化结构域样受体蛋白3抑制剂及其制备方法和应用
1.本发明属于医药技术领域,涉及一类1,2,3
‑
三氮唑类小分子化合物的制备方法和应用,具体地涉及一种新的能够抑制炎症小体核苷酸结合寡聚化结构域样受体蛋白3(nlrp3)和白介素
‑
1β(il
‑
1β)的小分子药物,所述化合物的制备方法,所述化合物作为炎症小体核苷酸结合寡聚化结构域样受体蛋白抑制剂或者白介素
‑
1β抑制剂药物在制备抗炎症药物以及自身免疫病药物中的应用。
背景技术:
2.下面关于与本发明相关的背景介绍用于帮助对本发明的理解,但不应被认为是本发明的已有技术。所有引用的出版物都被全文参考。
3.众所周知,炎症和免疫与多种疾病的发生发展密切相关,如sars
‑
cov
‑
2 引起的致命性的细胞因子风暴可导致多器官功能衰竭和死亡(filippou andkaragiannis,2020)。许多自身免疫性疾病是由白介素
‑
1β(il
‑
1β)、肿瘤坏死因子(tnf
‑
α)、白介素
‑
6(il
‑
6)等炎症因子的内稳态紊乱引起的,先天免疫与适应性免疫的相互作用最终会导致机体组织损伤(vernino,2020,berthelot andsibilia,2019)。在银屑病的发病机制中,tnf
‑
α起着核心作用(boehncke and2015);cryopyrin相关周期性综合征(caps)是一种罕见的遗传性自身炎症性疾病,以全身、皮肤、肌肉骨骼和中枢神经系统炎症为特征,髓源的促炎细胞因子的过度释放是引起全身和组织炎症导致疾病症状的关键(booshehriand hoffman,2019)。
4.炎症小体核苷酸结合寡聚化结构域样受体蛋白3(nlrp3)是这些过程中的关键调节因子,也是抗炎和其他相关疾病的重要药物靶蛋白(green et al.,2018, dinarello,2018)。nlrp3被一些病原体和损伤相关分子(pamps和damps)激活后,促进半胱天冬酶
‑
1前体(pro
‑
caspase
‑
1)的自我剪切,从而加速il
‑
1β前体的剪切和il
‑
1β的胞外释放(hoyle et al.,2020,swanson et al.,2019)。目前,这些自身免疫性疾病的临床治疗大多采用抗对应细胞因子的单克隆抗体。然而,单克隆抗体制剂属于生物大分子制剂,具有开发难度大,运输和储存成本高的缺点。此外,长期使用单抗制剂也可能引起机体抗药性或过敏等副作用。寻找有效的小分子来降低il
‑
1β或tnf
‑
α的安全性仍然是学术界的普遍目标(ecker et al.,2015, wolska
‑
washer and robak,2019)。
5.一些小分子具有抑制il
‑
1β或其他细胞因子释放的活性。例如mcc950,它在人类单核细胞中表现出出色的活性,ic50值为8.1nm,但在ii期临床试验中发现具有肝毒性(mangan et al.,2018,green et al.,2018)。因此,目前仍亟需寻找到一种安全有效的减轻nlrp3炎症的药物小分子。羧胺三唑 (carboxyamidotriazole,cai)作为一种口服广谱抗癌药物,在iii期临床试验中具有可信的安全性(bonnefond et al.,2018,das,2018,yang et al.,2008)(berlinet al.,2002,omuro et al.,2018,winters et al.,2005,hussain et al.,2003,si et al., 2020),在体外和体内均被确定为抑制nlrp3炎症体介导的il
‑
1β
释放的重要抑制剂(shi et al.,2019,chen et al.,2017,si et al.,2020,ju et al.,2012,guo et al., 2012)。除il
‑
1β外,cai的抗炎活性也表现为抑制tnf
‑
α等其他炎性细胞因子的分泌(guo et al.,2008,lu et al.,2020)。但cai在抑制il
‑
1β和tnf
‑
α释放方面的选择性较差(stephenson et al.,2020)。目前已经证实,tnf
‑
α抑制剂与免疫抑制的风险相关,这可能导致患者严重感染或肿瘤复发等严重副作用(singh etal.,2020,holmer and singh,2019,radner and aletaha,2015,askling et al.,2011)。因此,开发选择性的il
‑
1β小分子抑制剂作为抗炎药、治疗自身免疫病、提高此类药物的安全性具有重要意义。
技术实现要素:
6.本发明旨在针对现有技术的缺陷,提供一种新的能够抑制炎症小体核苷酸结合寡聚化结构域样受体蛋白3的小分子药物的制备方法和应用,以解决现有技术中缺乏一种类似化合物的技术问题;
7.本发明要解决的另一技术问题是发明背景中所述化合物的活性低、稳定性较差;
8.本发明要解决的又一技术问题是提供所述化合物作为炎症小体核苷酸结合寡聚化结构域样受体蛋白3抑制剂和白介素
‑
1β抑制剂在抗炎症药物以及自身免疫病药物中的应用。
9.本发明提供了一类式i所述的化合物及其药学上可用的盐:
[0010][0011]
其中:
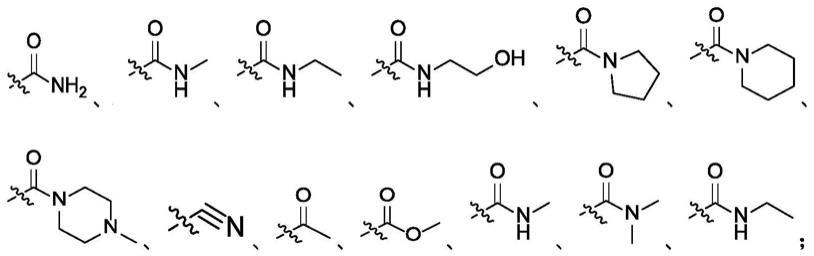
[0012]
r1独立地选自:酰胺基、氰基、酯基、酰基、取代酰基、取代酰胺基;
[0013]
r2独立地选自:氢、氨基、烷基化氨基、
[0014]
其中,r3独立地选自:氢、c1‑
c6烷酰基、环丙酰基、糠酰基、四氢糠酰基、苯甲酰基、3
‑
溴丙酰胺基、4
‑
溴丁酰胺基;
[0015]
r4独立地选自:氢、c1‑
c6烷酰基、环丙酰基、糠酰基、四氢糠酰基、苯甲酰基、3
‑
溴丙酰胺基、4
‑
溴丁酰胺基。
[0016]
进一步地,优选如下:
[0017]
r1独立地选自:
[0018][0019]
r2独立地选自:
[0020][0021]
更进一步地,优选下列化合物及其药学上可用的盐:
[0022]
[0023]
[0024][0025]
本发明提供了一种药物组合物,包含上述化合物或其药学上可用的盐,以及药学上可接受的载体,包括稀释剂。
[0026]
同时,本发明提供了上述化合物或其药学上可用的盐、上述药物组合物在制备炎症小体核苷酸结合寡聚化结构域样受体蛋白3小分子抑制剂、白介素
‑
1 β与肿瘤坏死因子
‑
α抑制剂药物的应用。
[0027]
作为优选,上述化合物或其药学上可用的盐、上述药物组合物是针对抗肿瘤或者肿瘤放化疗增敏的药物。
[0028]
同时,本发明提供了上述化合物的制备方法,包括以下步骤:
[0029]
1)由羧胺三唑在极性溶剂中与碘甲烷及氢化钠反应得到化合物a;
[0030]
2)由羧胺三唑在极性溶剂中与对氯苯磺酸乙酯及氢化钠反应得到化合物a;
[0031]
3)由化合物b与碳酸钾以及氰基化合物反应得到化合物c;
[0032]
4)由化合物c在极性溶剂中与碘甲烷及氢化钠反应得到化合物d;
[0033]
5)由化合物c在极性溶剂中与对氯苯磺酸乙酯及氢化钠反应得到化合物d;
[0034]
6)由羧胺三唑与酰氯在浓硫酸催化下下得到化合物e;
[0035]
7)由化合物b与丙炔酰胺在五水合硫酸铜、抗坏血酸钠的催化下在极性混合溶剂中反应得到化合物f。
[0036]
其中化合物结构如下:
[0037][0038]
作为优选,步骤1)2)4)5)所述反应是在氮气保护、加热条件下进行的。
[0039]
作为优选,步骤6)所述反应是在低温下进行的。更优的,反应温度为0摄氏度。
[0040]
作为优选,步骤7)所述反应在极性混合溶剂中进行的,为体积比为1∶2∶1的二氯甲烷
‑
叔丁醇
‑
水的混合溶剂。
[0041]
作为优选,步骤3)所述反应在碱性条件下进行,碱包括但不限于醋酸钠、醋酸钾、甲醇钠、乙醇钠、甲醇钾、乙醇钾、氟化钠、氟化钾、碳酸钾、碳酸钠、碳酸氢钠、碳酸铯。更优的,为碳酸钾。
[0042]
作为优选,步骤1)2)4)5)所述反应在极性溶剂中进行的,包括但不限于n,n
‑
二甲基甲酰胺、二甲基亚砜、乙腈、丙酮、甲基乙基酮、1,4
‑
二氧六环、叔丁醇、水。更优的,为n,n
‑
二甲基甲酰胺。
[0043]
作为优选,步骤7)所述反应在混合极性溶剂中进行的,包括但不限于 n,n
‑
二甲基甲酰胺、二甲基亚砜、叔丁醇乙腈、丙酮、甲基乙基酮、1,4
‑
二氧六环、水。更优的,为水/二氯甲烷/叔丁醇。
[0044]
以上技术方案的相关技术术语,除非特殊解释,否则将遵循下面的定义。
[0045]
术语“烷基”指具有指定碳原子数的直链或支链烃基,因此例如,在此使用的术语“c1‑
c6烷基”是指具有至少1个且至多6个的烷基。本发明中所使用的此类支链或直链烷基的实例包括,但不限于,甲基、乙基、正丙基、异丙基、异丁基、正丁基、叔丁基、正戊基、异戊基、正己基等。
[0046]
术语“随意取代”或“取代”指基团带有1
‑
2个不同的取代基,可以分别是低碳数烷基、低碳数芳基、低碳数芳烷基、低碳数环状烷基、低碳数杂环烷基、羟基、低碳数烷氧基、低碳数芳氧基、多卤代烷氧基、芳烷氧基、低碳数杂芳基、低碳数杂芳环氧基、低碳数杂芳烷基、低碳数杂芳烷氧基、叠氮基、氨基、卤素、低碳数烷巯基、氧基、低碳数酰烷基、低碳数羧酸酯基、羧酸、酰胺基、硝基、低碳数酰氧基、低碳数胺烷基、低碳数烷胺芳基、低碳数烷芳基、低碳数烷胺烷基、低碳数烷氧芳基、低碳数芳胺基、低碳数芳烷胺基、磺酰基、低碳数酰胺烷芳基、低碳数酰胺芳基、低碳数羟烷基、低碳数卤代烷基、低碳数烷胺烷酸基、低碳数脲烷基、氰基、低碳数烷氧烷基、低碳数多卤代烷基、低碳数芳烷氧烷基。
具体实施方式
[0047]
本发明中的化合物及制备可以通过下面的实例更好地说明。这些实例不应被解读为本发明的局限,现在已知的或将来开发的这些化合物的变化体也应被认为属于本发明的范畴并申请保护。
[0048]
以下将对本发明的具体实施方式进行详细描述。为了避免过多不必要的细节,在一下实施例中对属于公知的结构或功能将不进行详细描述。
[0049]
以下实施例中所使用的近似性语言可用于定量表述,表明在不改变基本功能的情况下可允许数量有一定的变动。因此,用“大约”、“左右”等语言所修正的数值不限于该准确数值本身。在一些些实施例中,“大约”表示允许其修正的数值在正负百分之十(
±
10%)的范围内变化,比如,“大约100”表示的是可以是90到110之间的任何数值。此外,在“大约第一数值到第二数值”的表述中,大约同时修正第一和第二数值两个数值。在某些情况下,近似性语言可能与测量仪器的精度有关。
[0050]
除有定义外,以下实施例中所使用的技术和科学术语具有与本发明所属领域技术人员普遍理解的相同含义。
[0051]
实施例11
‑
(3,5
‑
二氯
‑4‑
(4
‑
氯苯甲酰基)苄基)
‑5‑
(甲胺基)
‑
1h
‑
1,2,3
‑
三唑
ꢀ‑4‑
甲酰胺(a1)
[0052][0053]
在氮气保护下,将羧胺三唑(300mg,0.71mmol),氢化钠(34mg,0.85mmol) 和碘甲烷(151mg,1.06mmol)加入n,n
‑
二甲基甲酰胺中。反应液于60℃搅拌4 小时。停止反应,冷至室温,加入5ml乙酸乙酯,水洗三次,乙酸乙酯层以无水硫酸钠干燥,过滤,在减压条件下蒸干,残余物通过柱色谱分离(洗脱剂石油醚∶乙酸乙酯1∶1),得白色固体(0.241g,77.7%)。1h nmr(400mhz, dmso
‑
d6)δ7.78(d,j=8.6hz,2h),7.68(d,j=8.7hz,2h),7.62(s,1h),7.39(s, 2h),7.22(s,1h),6.51(q,j=5.4hz,1h),5.70(s,2h),2.97(d,j=5.4hz,3h).
[0054]
参照实施例1的步骤,可以制备如下化合物:
[0055][0056]1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑5‑
(dimethylamino)
‑
1h
‑
1,2,3
‑
triazole
‑4‑ꢀ
carboxamide(a2)
[0057]1h nmr(400mhz,dmso
‑
d6)δ7.84(s,1h),7.78(d,j=8.7hz,2h),7.68(d,j= 8.7hz,2h),7.53(s,2h),7.48(s,1h),5.59(s,2h),2.72(s,6h).
[0058]
实施例2 1
‑
(3,5
‑
二氯
‑4‑
(4
‑
氯苯甲酰基)苄基)
‑5‑
(乙氨基)
‑
1h
‑
1,2,3
‑
三唑
ꢀ‑4‑
甲酰胺(a3)
[0059][0060]
在氮气保护下,将羧胺三唑(425mg,1.0mmol),氢化钠(48mg,1.2mmol)和对氯苯磺酸乙酯(300mg,1.5mmol)加入n,n
‑
二甲基甲酰胺中。反应液于室温搅拌12小时。停止反应,加入5ml乙酸乙酯,水洗三次,乙酸乙酯层以无水硫酸钠干燥,过滤,在减压条件下蒸干,残余物通过柱色谱分离(洗脱剂石油醚∶乙酸乙酯2∶1),得白色固体(0.309g,68.5%)。1h nmr(400mhz,dmso
‑
d6)δ7.77 (d,j=8.6hz,2h),7.68(d,j=8.7hz,2h),7.66(s,1h),7.40(s,2h),7.24(s,1h), 6.33(t,j=6.3hz,1h),5.66(s,2h),3.42(p,j=7.0hz,2h),1.02(t,j=7.1hz,3h).
[0061]
实施例3 5
‑
氨基
‑1‑
(3,5
‑
二氯
‑4‑
(4
‑
氯苯甲酰基)苄基)
‑
n
‑
甲基
‑
ih
‑
1,2,3
‑
三唑
‑4‑
甲酰胺(c1)
[0062][0063]
将(4
‑
(叠氮甲基)
‑
2,6
‑
二氯苯基)(4
‑
氯苯基)甲酮(681mg,2mmol)、 2
‑
氰基
‑
n
‑
甲基乙酰胺(255mg,2.6mmol)和碳酸钾(1161mg,8.4mmol)在二甲基亚砜(1ml)中搅拌过夜制得。反应液中加入5ml乙酸乙酯,水洗三次,乙酸乙酯层以无水硫酸钠干燥,过滤,在减压条件下蒸干,残余物通过柱色谱分离(洗脱剂石油醚∶乙酸乙酯2∶1),得白色固体(0.723g,82.7%)。1h nmr(400 mhz,dmso
‑
d6)δ8.11(q,j=4.8hz,1h),7.78(d,j=8.6hz,2h),7.67(d,j=8.6 hz,2h),7.46(s,2h),6.51(s,2h),5.54(s,2h),2.74(d,j=4.7hz,3h)。
[0064]
参照实施例2的步骤,可以制备如下化合物:
[0065][0066]5‑
amino
‑1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑
n,n
‑
dimethyl
‑
1h
‑
1,2,3
‑ꢀ
triazole
‑4‑
carboxamide
[0067]1h nmr(400mhz,dmso
‑
d6)δ7.77(d,j=8.6hz,2h),7.68(d,j=8.5hz,2h), 7.47(s,2h),6.72(s,2h),5.54(s,2h),2.97(s,3h).
[0068][0069]5‑
amino
‑1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑
n
‑
ethyl
‑
1h
‑
1,2,3
‑
triazole
‑ꢀ4‑
carboxamide
[0070]1h nmr(400mhz,dmso
‑
d6)δ8.18(t,j=5.9hz,1h),7.76(d,j=8.6hz,2h), 7.66(d,j=8.6hz,2h),7.45(s,2h),6.51(s,2h),5.52(s,2h),3.23(p,j=7.0hz, 2h),1.08(t,j=7.1hz,3h).
[0071][0072]5‑
amino
‑1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑
n
‑
(2
‑
hydroxyethyl)
‑
1a
‑
1,2,3
‑ꢀ
triazole
‑4‑
carboxamide
[0073]1h nmr(400mhz,dmso
‑
d6)δ8.01(t,j=5.8hz,1h),7.77(d,j=8.6hz,2h),7.67(d,j=8.6hz,2h),7.46(s,2h),6.54(s,2h),5.54(s,2h),4.76(t,j=5.5hz, 1h),3.48(q,j=6.1hz,2h),3.30(q,j=6.1hz,2h).
[0074][0075]
(5
‑
amino
‑1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑
1h
‑
1,2,3
‑
triazol
‑4‑ꢀ
yl)(pyrrolidin
‑1‑
yl)methanone
[0076]1h nmr(400mhz,dmso
‑
d6)δ7.77(d,j=8.7hz,2h),7.67(d,j=8.7hz,2h), 7.47(s,2h),6.72(s,2h),5.55(s,2h),3.97(t,j=6.8hz,2h),3.47(t,j=6.9hz, 2h),1.93(p,j=6.8hz,2h),1.80(p,j=6.7hz,2h).
[0077][0078]
(5
‑
amino
‑1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑
1h
‑
1,2,3
‑
triazol
‑4‑ꢀ
yl)(piperidin
‑1‑
yl)methanone
[0079]1h nmr(400mhz,dmso
‑
d6)δ7.77(d,j=8.7hz,2h),7.68(d,j=8.8hz,2h), 7.49(s,2h),6.71(s,2h),5.54(s,2h),4.29(s,2h),3.57(s,2h),1.64(s,2h),1.54(s, 4h).
[0080][0081]
(5
‑
amino
‑1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑
1h
‑
1,2,3
‑
triazo1
‑4‑
y1)(4
‑ꢀ
methylpiperazin
‑1‑
yl)methanone
[0082]1h nmr(400mhz,dmso
‑
d6)δ7.77(d,j=8.4hz,2h),7.68(d,j=8.4hz,2h), 7.49(s,2h),6.75(s,2h),5.55(s,2h),4.35(s,2h),3.60(s,2h),2.36(s,4h),2.20(s, 3h).
[0083][0084]5‑
amino
‑1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑
1h
‑
1,2,3
‑
triazole
‑4‑
carbonitrile
[0085]1h nmr(400mhz,dmso
‑
d6)δ7.78(d,j=8.6hz,2h),7.68(d,j=8.6hz,2h), 7.51(s,2h),7.34(s,2h),5.52(s,2h).
[0086][0087]1‑
(5
‑
amino
‑1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑
1h
‑
1,2,3
‑
triazol
‑4‑
y1)ethan
‑ꢀ1‑
one
[0088]1h nmr(400mhz,dmso
‑
d6)δ7.79(d,j=8.6hz,2h),7.68(d,j=8.6hz,2h), 7.59(s,2h),5.78(s,2h),2.51(s,3h).
[0089][0090]
methyl 5
‑
amino
‑1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑
1h
‑
1,2,3
‑
triazole
‑4‑ꢀ
carboxylate
[0091]1h nmr(400mhz,dmso
‑
d6)δ7.77(d,j=8.6hz,2h),7.67(d,j=8.6hz,2h), 7.47(s,2h),6.80(s,2h),5.55(s,2h),3.79(s,3h).
[0092]
实施例4 1
‑
(3,5
‑
二氯
‑4‑
(4
‑
氯苯甲酰基)苄基)
‑
n
‑
甲基
‑5‑
(甲胺基)
‑
1h
‑ꢀ
1,2,3
‑
三唑
‑4‑
甲酰胺(d1)
[0093][0094]
在氮气保护下,将羧胺三唑5
‑
氨基
‑1‑
(3,5
‑
二氯
‑4‑
(4
‑
氯苯甲酰基)苄基)
ꢀ‑
n
‑
甲基
‑
1h
‑
1,2,3
‑
三唑
‑4‑
羧酰胺(300mg,0.69mmol),氢化钠(33mg,0.82mmol) 和碘甲烷(147mg,1.04mmol)加入n,n
‑
二甲基甲酰胺中。反应液于60℃搅拌4 小时。停止反应,冷至室温,加入5ml乙酸乙酯,水洗三次,乙酸乙酯层以无水硫酸钠干燥,过滤,在减压条件下蒸干,残余物通过柱色谱分离(洗脱剂石油醚∶乙酸乙酯1∶1),得白色固体(0.340g,75.3%)。1h nmr(400mhz,dmso
‑ꢀ
d6)δ8.21(q,j=4.6hz,1h),7.78(d,j=8.6hz,2h),7.68(d,j=8.6hz,2h),7.39 (s,2h),6.44(q,j=5.4hz,1h),5.68(s,2h),2.97(d,j=5.4hz,3h),2.74(d,j=4.7 hz,3h).
[0095]
参照实施例4的步骤,可以制备如下化合物:
[0096][0097]1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑5‑
(dimethylamino)
‑
n
‑
methyl
‑
1h
‑
1,2,3
‑ꢀ
triazole
‑4‑
carboxamide
[0098]1h nmr(400mhz,dmso
‑
d6)δ8.48(q,j=4.5hz,1h),7.77(d,j=8.6hz, 2h),7.68(d,j=8.6hz,2h),7.52(s,2h),5.59(s,2h),2.77(d,j=4.7hz,3h),2.72 (s,6h).
[0099][0100]1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑
n,n
‑
dimethyl
‑5‑
(methylamino)
‑
1h
‑ꢀ
1,2,3
‑
triazole
‑4‑
carboxamide
[0101]1h nmr(400mhz,dmso
‑
d6)δ7.78(d,j=8.7hz,2h),7.68(d,j=8.7hz, 2h),7.42(s,2h),6.64(q,j=5.2hz,1h),5.62(s,2h),3.29(s,3h),2.98(s,3h),2.82(d,j=5.2hz,3h).
[0102][0103]1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑
n
‑
ethyl
‑5‑
(methylamino)
‑
1h
‑
1,2,3
‑ꢀ
triazole
‑4‑
carboxamide
[0104]1h nmr(400mhz,dmso
‑
d6)δ8.28(t,j=5.9hz,1h),7.78(d,j=8.6hz,2h), 7.68(d,j=8.7hz,2h),7.39(s,2h),6.46(q,j=5.4hz,1h),5.69(s,2h),3.24(qd, j=7.1,5.8hz,2h),2.97(d,j=5.4hz,3h),1.09(t,j=7.2hz,3h).
[0105]
实施例5 1
‑
(3,5
‑
二氯
‑4‑
(4
‑
氯苯甲酰基)苄基)
‑5‑
(乙氨基)
‑
n
‑
甲基
‑
1h
‑ꢀ
1,2,3
‑
三唑
‑4‑
甲酰胺(d5)
[0106][0107]
在氮气保护下,将5
‑
氨基
‑1‑
(3,5
‑
二氯
‑4‑
(4
‑
氯苯甲酰基)苄基)
‑
n
‑
甲基
‑ꢀ
1h
‑
1,2,3
‑
三唑
‑4‑
羧酰胺(425mg,0.97mmol),氢化钠(47mg,1.2mmol)和对氯苯磺酸乙酯(291mg,1.5mmol)加入n,n
‑
二甲基甲酰胺中。反应液于室温搅拌12 小时。停止反应,加入5ml乙酸乙酯,水洗三次,乙酸乙酯层以无水硫酸钠干燥,过滤,在减压条件下蒸干,残余物通过柱色谱分离(洗脱剂石油醚∶乙酸乙酯2∶1),得白色固体(0.330g,71.1%)。1h nmr(400mhz,dmso
‑
d6)δ 7.77(d,j=8.6hz,2h),7.68(d,j=8.7hz,2h),7.66(s,1h),7.40(s,2h),7.24(s, 1h),6.33(t,j=6.3hz,1h),5.66(s,2h),3.42(p,j=7.0hz,2h),1.02(t,j=7.1 hz,3h).
[0108]
参照实施例5的步骤,可以制备如下化合物:
[0109][0110]1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑
n
‑
ethyl
‑5‑
(ethylamino)
‑
1h
‑
1,2,3
‑ꢀ
triazole
‑4‑
carboxamide
[0111]1h nmr(400mhz,dmso
‑
d6)δ8.31(t,j=5.9hz,1h),7.77(d,j=8.7hz,2h),7.68(d,j=8.7hz,2h),7.40(s,2h),6.29(t,j=6.3hz,1h),5.65(s,2h),3.41(p, j=7.0hz,2h),3.25(p,j=7.1hz,2h),1.09(t,j=7.1hz,3h),1.02(t,j=7.1hz, 3h).
[0112]
实施例6 1
‑
(3,5
‑
二氯
‑4‑
(4
‑
氯苯甲酰基)苄基)
‑5‑
丙酰胺
‑
1h
‑
1,2,3
‑
三唑
‑4‑ꢀ
甲酰胺(e1)
[0113][0114]
将cai(300mg,0.71mmol)分散于2.5ml丙酰氯中,冰浴搅拌半小时。滴加2滴浓硫酸,继续冰浴搅拌。半小时后,撤去冰浴,室温搅拌过夜。停止反应,加入饱和碳酸氢钠溶液至ph中性。加入5ml乙酸乙酯,水洗三次,乙酸乙酯层以无水硫酸钠干燥,过滤,在减压条件下蒸干,残余物通过柱色谱分离(洗脱剂石油醚∶乙酸乙酯2∶1),得白色固体(0.364g,76.0%)。1h nmr(400mhz, dmso
‑
d6)δ10.21(s,1h),7.93(s,1h),7.77(d,j=8.3hz,2h),7.68(d,j=8.3hz, 2h),7.52(d,j=22.2hz,3h),5.56(s,2h),2.38(dd,j=7.6hz,2h),1.06(t,j=7.6 hz,3h).
[0115][0116]1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑5‑
(n
‑
propionylpropionamido)
‑
1h
‑
1,2,3
‑ꢀ
triazole
‑4‑
carboxamide
[0117]1h nmr(400mhz,dmso
‑
d6)δ10.69(s,1h),10.42(s,1h),7.76(d,j=8.7hz,2h), 7.69(d,j=8.7hz,2h),7.51(s,2h),5.63(s,2h),2.67(q,j=7.4hz,2h),2.39(q,j =7.5hz,2h),1.07(t,j=7.5hz,3h),1.03(t,j=7.4hz,3h).
[0118][0119]5‑
butyramido
‑1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑
1h
‑
1,2,3
‑
triazole
‑4‑ꢀ
carboxamide
[0120]1h nmr(400mhz,dmso
‑
d6)δ10.23(s,1h),7.93(s,1h),7.77(d,j=8.6hz, 2h),7.68(d,j=8.7hz,2h),7.54(s,1h),7.48(s,2h),5.56(s,2h),2.33(t,j=7.3 hz,2h),1.59(sext,j=7.4hz,2h),0.91(t,j=7.4hz,3h).
[0121][0122]5‑
(n
‑
butyrylbutyramido)
‑1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑
1h
‑
1,2,3
‑ꢀ
triazole
‑4‑
carboxamide
[0123]1h nmr(400mhz,dmso
‑
d6)δ10.70(s,1h),10.44(s,1h),7.6(d,j=8.6hz, 2h),7.69(d,j=8.7hz,2h),7.49(s,2h),5.63(s,2h),1.24(m,14h).
[0124][0125]5‑
(3
‑
bromopropanamido)
‑1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑
1h
‑
1,2,3
‑ꢀ
triazole
‑4‑
carboxamide
[0126]1h nmr(400mhz,dmso
‑
d6)δ10.56(s,1h),7.94(s,1h),7.78(d,j=8.6hz,2h), 7.68(d,j=8.7hz,2h),7.57(s,1h),7.51(s,2h),5.55(s,2h),3.87(t,j=6.3hz, 2h),2.92(t,j=6.3hz,2h).
[0127][0128]5‑
(4
‑
bromobutanamido)
‑1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑
1h
‑
1,2,3
‑ꢀ
triazole
‑4‑
carboxamide
[0129]1h nmr(400mhz,dmso
‑
d6)δ10.32(s,1h),7.92(s,1h),7.77(d,j=8.6hz,2h), 7.68(d,j=8.7hz,2h),7.54(s,1h),7.50(s,2h),5.57(s,2h),3.71(t,j=6.6hz, 2h),2.54(t,j=6.6hz,2h),2.02(p,j=6.6hz,2h).
[0130][0131]1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzy1)
‑5‑
isobutyramido
‑
1h
‑
1,2,3
‑
triazole
‑4‑ꢀ
carboxamide
[0132]1h nmr(400mhz,dmso
‑
d6)δ10.16(s,1h),7.93(s,1h),7.77(d,j=8.6hz,2h),7.68(d,j=8.7hz,2h),7.46(s,2h),5.56(s,2h),2.65(hept,j=6.7hz,1h), 1.10(d,j=6.9hz,6h).
[0133][0134]1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑5‑
(n
‑
isobutyrylisobutyramido)
‑
1h
‑
1,2,3
‑ꢀ
triazole
‑4‑
carboxamide
[0135]1h nmr(400mhz,dmso
‑
d6)δ10.77(s,1h),10.37(s,1h),7.76(d,j=8.6hz, 2h),7.69(d,j=8.6hz,2h),7.48(s,2h),5.64(s,2h),3.07(hept,j=6.8hz,1h), 2.66(hept,j=6.8hz,1h),1.10(d,j=6.7hz,6h),1.08(d,j=6.7hz,6h).
[0136][0137]1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑5‑
pivalamido
‑
1h
‑
1,2,3
‑
triazole
‑4‑ꢀ
carboxamide
[0138]1h nmr(400mhz,dmso
‑
d6)δ9.66(s,1h),7.92(s,1h),7.77(d,j=8.6hz,2h), 7.68(d,j=8.7hz,2h),7.51(s,1h),7.45(s,2h),5.57(s,2h),1.19(s,9h).
[0139][0140]1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑5‑
(n
‑
pivaloylpivalamido)
‑
1h
‑
1,2,3
‑ꢀ
triazole
‑4‑
carboxamide
[0141]1h nmr(400mhz,dmso
‑
d6)δ10.20(s,1h),9.87(s,1h),7.75(d,j=8.7hz,2h),7.68(d,j=8.8hz,2h),7.47(s,2h),5.65(s,2h),1.24(s,9h),1.21(s,9h).
[0142][0143]1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑5‑
(2
‑
ethylbutanamido)
‑
1h
‑
1,2,3
‑ꢀ
triazole
‑4‑
carboxamide
[0144]1h nmr(400mhz,dmso
‑
d6)δ10.26(s,1h),7.92(s,1h),7.75(d,j=8.6hz,2h), 7.68(d,j=8.7hz,2h),7.51(s,1h),7.43(s,2h),5.57(s,2h),2.31(m,1h),1.57(m, 2h),1.45(m,2h),0.87(m,6h).
[0145][0146]1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑5‑
(2
‑
ethyl
‑
n
‑
(2
‑ꢀ
ethylbutanoyl)butanamido)
‑
1h
‑
1,2,3
‑
triazole
‑4‑
carboxamide
[0147]1h nmr(400mhz,dmso
‑
d6)δ10.86(s,1h),10.48(s,1h),7.75(d,j=8.8hz, 2h),7.69(d,j=8.9hz,2h),7.44(s,2h),5.66(s,2h),2.81(m,1h),2.31(m,1h), 1.57(m,4h),1.44(m,4h),0.86(m,12h).
[0148][0149]1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑5‑
(furan
‑2‑
carboxamido)
‑
1h
‑
1,2,3
‑ꢀ
triazole
‑4‑
carboxamide
[0150]1h nmr(400mhz,dmso
‑
d6)δ10.57(s,1h),8.02(dd,j=1.8,0.8hz,1h),7.98 (s,1h),7.76(d,j=8.7hz,2h),7.69(d,j=8.8hz,2h),7.58(s,1h),7.49(s,2h), 7.39(dd,j=3.6,0.8hz,1h),6.75(dd,j=3.5,1.7hz,1h),5.65(s,2h).
[0151][0152]5‑
benzamido
‑1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑
1h
‑
1,2,3
‑
triazole
‑4‑ꢀ
carboxamide
[0153]1h nmr(400mhz,dmso
‑
d6)δ10.63(s,1h),8.01(s,1h),7.96(d,j=7.4hz, 2h),7.75(d,j=8.6hz,2h),7.68(d,j=8.6hz,2h),7.64(d,j=7.4hz,1h),7.56 (m,3h),7.48(s,2h),5.68(s,2h).
[0154][0155]5‑
(cyclopropanecarboxamido)
‑1‑
(3,5
‑
dichloro
‑4‑
(4
‑
chlorobenzoyl)benzyl)
‑
1h
‑
1,2,3
‑ꢀ
triazole
‑4‑
carboxamide
[0156]1h nmr(400mhz,dmso
‑
d6)δ10.56(s,1h),7.95(s,1h),7.77(d,j=8.7hz, 2h),7.68(d,j=8.8hz,2h),7.57(s,1h),7.45(s,2h),5.55(s,2h),4.03(q,j=7.1 hz,1h),0.85(m,4h).
[0157][0158]5‑
(n
‑
(cyclopropanecarbonyl)cyclopropanecarboxamido)
‑1‑
(3,5
‑
dichloro
‑4‑
(4
‑ꢀ
chlorobenzoyl)benzyl)
‑
1h
‑
1,2,3
‑
triazole
‑4‑
carboxamide
[0159]1h nmr(400mhz,dmso
‑
d6)δ11.11(s,1h),10.76(s,1h),7.77(d,j=8.6hz, 2h),7.69(d,j=8.7hz,2h),7.48(s,2h),5.64(s,2h),2.43(s,1h),1.88(s,1h), 0.87(m,8h).
[0160]
实施例7 1
‑
(3,5
‑
二氯
‑4‑
(4
‑
氯苯甲酰基)苄基)
‑
1h
‑
1,2,3
‑
三唑
‑4‑
甲酰胺 (f1)
[0161][0162]
将(4
‑
(叠氮甲基)
‑
2,6
‑
二氯苯基)(4
‑
氯苯基)甲酮(300mg,0.88mmol),丙炔酰胺(74mg,1.06mmol)与五水合硫酸铜(23mg,0.09mmol)和抗坏血酸钠(52mg,0.3mmol)在16ml的二氯甲烷/叔丁醇/水(1/2/1)v/v/v中制备。室温搅拌2小时后,停止反应。水洗三次,乙酸乙酯层以无水硫酸钠干燥,过滤,在减压条件下蒸干。通过柱色谱分离(洗脱剂石油醚∶乙酸乙酯1∶2),得白色固体(0.322g,89.7%)。1h nmr(400mhz,dmso
‑
d6)δ8.74(s,1h),7.95(s,1h), 7.79(d,j=8.6hz,2h),7.69(s,2h),7.67(d,j=8.7hz,2h),7.56(s,1h),5.77(s, 2h).
[0163]
实施例8 il
‑
1β含量elisa检测实验
[0164]
1.实验试剂:
[0165][0166]
2.实验步骤:
[0167]
2.1 bmdm细胞分离及培养
[0168]
2.1.1脱颈处死小鼠后,将小鼠浸没于75%酒精3
‑
5min;
[0169]
2.1.2在超净台内用剪刀和镊子分离小鼠腿部的皮肤和肌肉,分离出胫骨和股骨,将胫骨和股骨在75%酒精和只含双抗的培养基分别浸泡3min;
[0170]
2.1.3剪掉股骨和胫骨两端骨骺,用1ml注射器吸取无菌pbs冲洗骨髓到 50ml离心管中,每根骨头用5ml pbs冲洗;
[0171]
2.1.4用300目滤网过滤骨髓细胞悬液到50ml离心管。在4℃下1500g离心5min,弃去上清液,加入1ml红细胞裂解液裂解红细胞,2min后用3 ml bmdm培养基重悬细胞,再次离心;
[0172]
2.1.5加入10m1bmdm培养基重悬细胞,于10cm大皿在孵箱中孵育3小时;
[0173]
2.1.6收集未贴壁的骨髓细胞于15m1离心管中,离心后细胞计数,以1x105密度在96孔板中种板,第3天更换培养基,第6天可用于实验。
[0174]
2.2刺激炎症
[0175]
第6天早晨,以50ng/ml脂多糖溶液加入96孔板,刺激三小时。
[0176]
2.3给药
[0177]
将化合物以终浓度20μm加入每孔,孵育30分钟
[0178]
2.4 atp的刺激
[0179]
加入终浓度5mm atp溶液,进一步刺激nlrp3炎症。
[0180]
2.5取细胞上清液,按elisa试剂盒说明书操作,检测il
‑
1β含量。
[0181]
表一:化合物诱导白介素
‑
1β表达抑制率
[0182][0183]
[0184]
[0185]
[0186][0187]
elisa结果表明,除了化合物xix、xx、xxii、xxiv、xxviii、xxx、 xxxii、xxxiv外,其他化合物都能不同程度地对il
‑
1β产生抑制作用。其中化合物viii、xxv、xxxi的活性优于cai。
[0188]
比较各化合物活性,初步构效关系提示,r2进行单酰基化为此类化合物的活性结构。
[0189]
实施例9 tnf
‑
α含量elisa检测实验
[0190]
1.实验试剂:
[0191][0192]
2.实验步骤:
[0193]
2.1 raw264.7细胞培养
[0194]
将细胞以2x105密度在96孔板中种板,培养3小时。
[0195]
2.2给药
[0196]
将化合物以终浓度20μm加入每孔,孵育30分钟。
[0197]
2.3刺激炎症
[0198]
以终浓度50ng/ml脂多糖溶液刺激细胞三小时
[0199]
2.4取细胞上清液,按elisa试剂盒说明书操作,检测tnf
‑
α含量。
[0200]
表二:化合物诱导肿瘤坏死因子
‑
α表达抑制率
[0201][0202]
[0203]
[0204]
[0205][0206]
elisa结果表明,除了化合物xiv、xxiv外,其他化合物都能不同程度地对tnf
‑
α产生抑制作用。其中绝大多数化合物对tnf
‑
α的抑制小于cai,这有助于提高化合物对il
‑
1β的选择抑制作用。
[0207]
实施例10化合物对il
‑
1β的选择性
[0208]
表三:化合物对il
‑
1β的选择性指数(selectivity index)
[0209]
[0210][0211]
用化合物对il
‑
1β的抑制率与tnf
‑
α抑制率的比值来表示化合物对il
‑
1β的选择性。很多化合物的选择性都优于cai(0.82)。其中化合物xxvi、xxxi 的选择性指数高达十几,这表明化合物对il
‑
1β的选择性大大提高。
[0212]
实施例11代表性化合物对il
‑
1β抑制的ic
50
[0213]
表四:代表性化合物对il
‑
1β抑制的ic
s0
(μm)
[0214][0215]
结果表明,代表性化合物对il
‑
1β有着稳定的抑制活性。
[0216]
因此,式i所示的化合物均能不同程度地抑制il
‑
1β的释放。其中代表性化合物xxvi、xxxi在保持与cai相当的活性的同时,还极大地提高了对il
‑
1β抑制的选择性。式i所示的化合物有望成为抗炎新药或自身免疫病药物。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1