一种吡唑并嘧啶并三唑环类化合物的制备方法与流程
1.本发明属于有机化合物合成技术领域,具体涉及一种吡唑并嘧啶并三唑环类化合物的制备方法。
背景技术:
2.近年来中枢神经系统疾病的发病率和致死率呈上升趋势,严重影响患者的生活质量,给患者及社会带来极大负担。腺苷是一种分子式为c
10
h
13
n5o4的内源性嘌呤核苷,存在于每一个细胞中,其通过与特定的细胞表面受体相互作用而调节广泛的生理功能,这些受体被分类为a1、a
2a
、a
2b
和a3腺苷受体亚型。其中,a
2a
受体在治疗阿尔茨海默病、帕金森病、亨廷顿病等中枢神经系统疾病中发挥重要作用,包括调控胶质细胞炎症反应、神经元可塑性及屏障通透性等,因此发展高选择性的a
2a
受体拮抗剂具有重要意义,其中吡唑并嘧啶并三唑环是应用较为广泛的a
2a
受体拮抗剂。
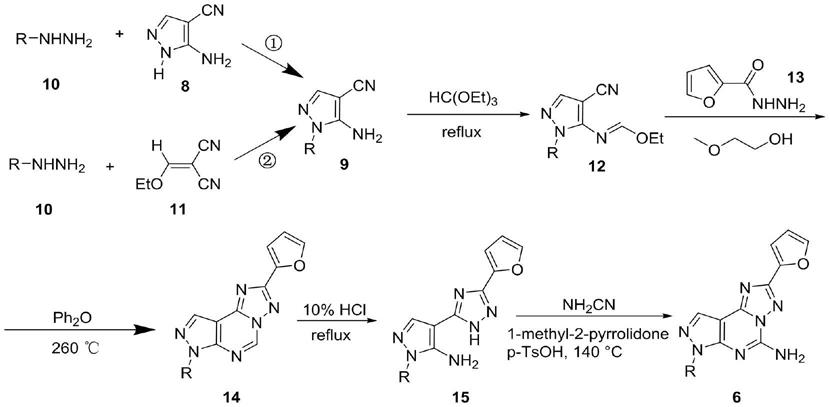
3.现有的合成吡唑并嘧啶并三唑环的路径如下(pier giovanni baraldi et al.,pyrazolo[4,3
‑
e]
‑
1,2,4
‑
triazolo[1,5
‑
c]pyrimidine derivatives:potent and selective a2a adenosine antagonists,j.med.chem.1996,39,1164
‑
1171):
[0004][0005]
在合成n1‑
烷基
‑4‑
氰基
‑5‑
氨基吡唑9的基础上,利用氨基和原甲酸三乙酯发生缩合得到乙氧基亚胺12,而后其再与呋喃甲酰肼13缩合成环得到14,14中的二氢嘧啶环水解重新回到氨基吡唑环,即得到15,最后15中的三唑n进攻氰胺进而与吡唑环发生分子内缩合成环,最终得到产物6。
[0006]
现有的合成吡唑并嘧啶并三唑环的制备步骤繁杂、不易工业化生产。
技术实现要素:
[0007]
有鉴于此,本发明提供了一种吡唑并嘧啶并三唑环类化合物的制备方法。本发明提供的制备方法步骤简单,易工业化生产。
[0008]
为了解决上述技术问题,本发明提供了吡唑并嘧啶并三唑环类化合物的制备方
法,包括以下步骤:
[0009]
将2
‑
氨基
‑
4,6
‑
二氯嘧啶
‑5‑
甲醛与式i所示结构的肼化合物混合进行缩合反应,得到式ii所示结构的吡啶并吡唑;
[0010]
将所述式ii所示结构的吡啶并吡唑与式iii所示结构的酰基肼混合进行取代反应,得到式iv所示结构的4
‑
酰肼基吡啶并吡唑;
[0011]
将所述式iv所示结构的4
‑
酰肼基吡啶并吡唑和缩合剂混合进行缩合重排反应,得到式v所示结构的吡唑并嘧啶并三唑环类化合物;
[0012][0013]
所述式i~v中,r1为丁基、异戊基、苯乙基、甲基或苯基,r2为呋喃基、烷基、苯基或吡啶基。
[0014]
优选地,所述2
‑
氨基
‑
4,6
‑
二氯嘧啶
‑5‑
甲醛和式i所示结构的肼化合物的摩尔比为1:(1~3)。
[0015]
优选地,所述缩合反应的时间为12~24h。
[0016]
优选地,所述式ii所示结构的吡啶并吡唑和式iii所示结构的酰基肼的摩尔比为1:(1~5)。
[0017]
优选地,所述取代反应的温度为60~100℃,时间为12~20h。
[0018]
优选地,所述式iv所示结构的4
‑
酰肼基吡啶并吡唑和缩合剂的用量比为1g:(5~15)ml。
[0019]
优选地,所述缩合重排反应的温度为80~140℃,时间为8~24h。
[0020]
优选地,所述缩合剂包括n,n
‑
碳酰二咪唑、1
‑
(3
‑
二甲胺基丙基)
‑3‑
乙基碳二亚胺、2
‑
(7
‑
氮杂苯并三氮唑)
‑
n,n,n',n'
‑
四甲基脲六氟磷酸酯或o
‑
苯并三氮唑
‑
四甲基脲六氟磷酸盐。
[0021]
优选地,所述式i~v中,r1为丁基、异戊基或苯乙基,r2为呋喃基、吡啶基或苯基。
[0022]
优选地,所述式i所示结构的肼化合物由水合肼与卤代烃进行取代反应制得。
[0023]
本发明提供了一种吡唑并嘧啶并三唑环类化合物的制备方法,包括以下步骤:将2
‑
氨基
‑
4,6
‑
二氯嘧啶
‑5‑
甲醛与式i所示结构的肼化合物混合进行缩合反应,得到式ii所示结构的吡啶并吡唑;将所述式ii所示结构的吡啶并吡唑、式iii所示结构的酰基肼混合进行取代反应,得到式iv所示结构的4
‑
酰肼基吡啶并吡唑;将所述式iv所示结构的4
‑
酰肼基吡啶并吡唑和缩合剂混合进行缩合重排反应,得到式v所示结构的吡唑并嘧啶并三唑环类化合物。本发明提供的制备方法合成路径短,易于操作,适合工业化生产。
附图说明
[0024]
图1为实施例1制备得到的5
‑
氨基
‑2‑
呋喃基
‑7‑
苯乙基
‑
7h
‑
吡唑并[4,3
‑
e][1,2,4]三唑并[1,5
‑
c]嘧啶的核磁共振氢谱图;
[0025]
图2为实施例1制备得到的5
‑
氨基
‑2‑
呋喃基
‑7‑
苯乙基
‑
7h
‑
吡唑并[4,3
‑
e][1,2,4]三唑并[1,5
‑
c]嘧啶的核磁共振碳谱图。
具体实施方式
[0026]
本发明提供了一种吡唑并嘧啶并三唑环类化合物的制备方法,包括以下步骤:
[0027]
将2
‑
氨基
‑
4,6
‑
二氯嘧啶
‑5‑
甲醛与式i所示结构的肼化合物混合进行缩合反应,得到式ii所示结构的吡啶并吡唑;
[0028]
将所述式ii所示结构的吡啶并吡唑、式iii所示结构的酰基肼混合进行取代反应,得到式iv所示结构的4
‑
酰肼基吡啶并吡唑;
[0029]
将所述式iv所示结构的4
‑
酰肼基吡啶并吡唑和缩合剂混合进行缩合重排反应,得到式v所示结构的吡唑并嘧啶并三唑环类化合物;
[0030][0031]
所述式i~v中,r1为丁基、异戊基、苯乙基、甲基或苯基,r2为呋喃基、烷基、苯基或吡啶基。
[0032]
在本发明中,若无特殊说明,使用的原料均为本领域市售商品。
[0033]
本发明将2
‑
氨基
‑
4,6
‑
二氯嘧啶
‑5‑
甲醛与式i所示结构的肼化合物混合进行缩合反应,得到式ii所示结构的吡啶并吡唑。
[0034]
在本发明中,所述式i中,r1优选为苯乙基。
[0035]
在本发明中,所述2
‑
氨基
‑
4,6
‑
二氯嘧啶
‑5‑
甲醛和式i所示结构的肼化合物的摩尔比优选为1:(1~3)。
[0036]
在本发明中,所述缩合反应的温度优选为20~30℃,更优选为23~28℃;所述缩合反应的时间优选为12~24h,更优选为16~20h。
[0037]
在本发明中,所述缩合反应优选在有机溶剂中进行,所述有机溶剂优选为四氢呋喃。
[0038]
在本发明中,所述缩合反应优选三乙胺存在的条件下进行,所述2
‑
氨基
‑
4,6
‑
二氯
‑
嘧啶
‑5‑
甲醛与三乙胺的质量体积比优选为1g:(3~7)ml。
[0039]
在本发明中,所述三乙胺和有机溶剂的体积比优选为1:(5~7),更优选为1:6。
[0040]
在本发明中,所述2
‑
氨基
‑
4,6
‑
二氯嘧啶
‑5‑
甲醛、式i所示结构的、三乙胺和有机溶剂的混合优选包括以下步骤:
[0041]
将所述2
‑
氨基
‑
4,6
‑
二氯嘧啶
‑5‑
甲醛溶解于部分有机溶剂中,得到第一溶液;
[0042]
将所述第一溶液和三乙胺混合,得到第二溶液;
[0043]
将所述式i所示结构的肼化合物溶解于剩余有机溶剂中,得到肼化合物溶液;
[0044]
将所述肼化合物溶液滴加至所述第二溶液中,得到反应液。
[0045]
在本发明中,所述部分有机溶剂占有机溶剂的体积百分比优选为80~85%,更优选为83~84%。在本发明中,所述溶解的温度优选为1~9℃,更优选为3~7℃。
[0046]
在本发明中,所述滴加的速率优选为2~5滴/min,更优选为2~4滴/min。在本发明的实施例中,所述滴加优选在15min内完成。
[0047]
在本发明中,所述缩合反应完成后优选还包括将所得缩合反应产物依次进行旋蒸和层析分离。
[0048]
本发明对所述旋蒸的温度和时间没有特殊的限定,能够除去缩合反应后体系中的溶剂即可。
[0049]
在本发明中,所述层析分离用溶剂优选为石油醚和乙酸乙酯的混合物,所述石油醚和乙酸乙酯的质量比优选为1.8~5.2:1,更优选为5:1。
[0050]
在本发明中,以苯乙基肼为例,所述缩合反应的原理如式a所示:
[0051][0052]
在本发明中,所述式i所示结构的肼化合物优选由水合肼与卤代烃进行取代反应制得。本发明对所述水合肼、卤代烃以及取代反应的具体条件没有特殊的限定,采用本领域技术人员熟知的市售商品即可。在本发明中的具体实施例中,所述卤代烃优选包括卤代丁烷、卤代异戊烷、卤代甲烷或卤代烷基苯,所述卤代丁烷优选包括溴代丁烷或氯代丁烷,更优选为溴代丁烷;所述卤代异戊烷优选包括溴代异戊烷或氯代异戊烷,更优选为溴代异戊烷;所述卤代甲烷优选包括一溴甲烷或一氯甲烷,更优选为一溴甲烷;所述卤代烷基苯优选包括溴代甲基苯、氯代甲基苯、2
‑
氯乙基苯或2
‑
溴乙基苯,更优选为2
‑
溴乙基苯。
[0053]
在本发明中,所述水合肼和卤代烃的摩尔比优选为1:(1~3)。
[0054]
在本发明中,所述取代反应优选在乙醇中进行。
[0055]
在本发明中,所述水合肼和乙醇的体积比优选为(12.3~13.5):100,更优选为(12.6~13.1):100。
[0056]
在本发明中,所述水合肼、卤代烃和乙醇的混合优选包括以下步骤:
[0057]
将水合肼溶解于乙醇中,得到水合肼乙醇溶液;
[0058]
将卤代烃添加至所述水合肼乙醇溶液中。
[0059]
本发明将水合肼溶解于乙醇中,得到水合肼乙醇溶液。本发明对所述溶解无特殊限定。
[0060]
得到水合肼乙醇溶液后,本发明将卤代烃添加至所述水合肼乙醇溶液中。在本发明中,所述添加优选在回流的条件下进行。
[0061]
在本发明中,所述取代反应优选在回流的条件下进行,所述取代反应的温度优选为58~62℃,更优选为60℃;所述取代反应的时间优选为1~2h,更优选为1.5~1.8h。
[0062]
在本发明中,所述取代反应后优选还包括:将所得取代反应后产物依次进行旋蒸、萃取和除溶剂,得到所述式i所示结构的肼化合物。
[0063]
本发明对所述旋蒸的温度和时间没有特殊的限定。
[0064]
在本发明中,所述萃取用萃取溶剂优选为乙酸乙酯,所述萃取的次数优选为2~4次,更优选为3次。
[0065]
本发明在萃取后将所得有机相合并后与无水碳酸钾混合,以除去有机相中的水。在本发明中,除水后优选还包括:将所得有机相和无水碳酸钾混合的体系进行过滤;利用水泵抽去滤液中的溶剂。本发明对所述过滤无特殊限定,采用本领域常规的方式即可。在本发明中,所述利用水泵抽去滤液中的溶剂时的温度优选为78~82℃,更优选为80℃;压力优选为0.093~1.097bar,更优选为0.095bar。
[0066]
在本发明中,以卤代物为溴代乙基苯为例,所述取代反应的原理如式b所示:
[0067][0068]
得到式ii所示结构的吡啶并吡唑后,本发明将所述式ii所示结构的吡啶并吡唑、式iii所示结构的酰基肼混合进行取代反应,得到式iv所示结构的4
‑
酰肼基吡啶并吡唑。
[0069]
在本发明中,所述式iii中,r2优选为呋喃基。
[0070]
在本发明中,所述式ii所示结构的吡啶并吡唑和式iii所示结构的酰基肼的摩尔比优选为1:1~5,更优选为1:1.3~2。
[0071]
在本发明中,所述取代反应的温度优选为80~100℃,更优选为85~95℃,时间优选为12~20h,更优选为14~18h。
[0072]
在本发明中,所述取代反应优选在有机溶剂中进行,更优选为正丁醇。
[0073]
本发明对所述有机溶剂的用量无特殊限定,只要能够溶解完全即可。在本发明的实施例中,所述式ii所示结构的吡啶并吡唑与有机溶剂的质量体积比优选为2.67~2.79g:120ml。
[0074]
本发明对所述式ii所示结构的吡啶并吡唑、式iii所示结构的酰基肼和有机溶剂的混合无特殊限定,只要能够混合均匀即可。
[0075]
本发明优选采用tcl跟踪取代反应进程。
[0076]
在本发明中,所述取代反应后优选还包括:将所得取代反应溶液依次进行旋蒸和层析分离。
[0077]
本发明对所述旋蒸的温度和时间没有特殊的限定,能够除去取代反应后体系中的溶剂即可。
[0078]
在本发明中,所述层析分离用层析柱优选为硅胶柱,所述层析分离用溶剂优选为石油醚和乙酸乙酯的混合物,所述石油醚和乙酸乙酯的质量比优选为1~5:1,更优选为5:1。
[0079]
在本发明中,以2
‑
呋喃甲酰肼为例,所述取代反应的原理如式c所示:
[0080][0081]
得到式iv所示结构的4
‑
酰肼基吡啶并吡唑后,本发明将所述式iv所示结构的4
‑
酰肼基吡啶并吡唑和缩合剂混合进行缩合重排反应,得到式v所示结构的吡唑并嘧啶并三唑环类化合物。
[0082]
在本发明中,所述缩合剂优选包括n,n
‑
碳酰二咪唑(cdi)、1
‑
(3
‑
二甲胺基丙基)
‑3‑
乙基碳二亚胺(edci)、2
‑
(7
‑
氮杂苯并三氮唑)
‑
n,n,n',n'
‑
四甲基脲六氟磷酸酯(hatu)或o
‑
苯并三氮唑
‑
四甲基脲六氟磷酸盐(hbtu)。
[0083]
在本发明中,所述4
‑
酰肼基吡啶并吡唑和缩合剂的用量比优选为1.2~1.3g:10ml,更优选为1.25~1.28g:10ml。
[0084]
在本发明中,所述缩合重排反应的温度优选为100~140℃,更优选为110~130℃;所述缩合重排的时间优选为8~16h,更优选为10~14h。本发明优选采用tcl跟踪缩合重排反应进程。
[0085]
在本发明中,所述缩合重排反应后优选还包括:将所得缩合重排反应溶液依次进行旋蒸、层析分离和重结晶。
[0086]
本发明对所述旋蒸的温度和时间没有特殊的限定,能够除去过量的缩合剂即可。
[0087]
在本发明中,所述层析分离用溶剂优选为石油醚和乙酸乙酯的混合物,所述石油醚和乙酸乙酯的质量比优选为1~10:1,更优选为10:1。
[0088]
在本发明中,所述重结晶优选为将所得层析分离的产物和乙酸乙酯
‑
石油醚混合溶剂混合。
[0089]
在本发明中,以呋喃
‑2‑
羧酸n
’‑
(6
‑
氨基
‑1‑
苯乙基
‑
1h
‑
吡唑并[3,4
‑
d]嘧啶
‑
基)
‑4‑
酰肼为例,所述缩合重排反应的原理如式d所示:
[0090][0091]
本发明制备吡唑并嘧啶并三唑环类化合物的反应原理如式e所示:
[0092][0093]
本发明提供的制备方法中原料易得,合成路径短,易于工业化生产。
[0094]
为了进一步说明本发明,下面结合实施例对本发明提供的技术方案进行详细地描述,但不能将它们理解为对本发明保护范围的限定。
[0095]
实施例1
[0096]
本实施例中r1为苯乙基、r2为呋喃基;
[0097]
将12.3ml纯度为98%的水合肼溶解于100ml乙醇中,得到水合肼乙醇溶液;
[0098]
在回流的条件下将8.8g 2
‑
溴乙基苯滴加(15min滴加完成)至水合肼乙醇溶液中,回流条件下60℃进行第一取代反应1h;将第一取代反应后溶液旋蒸;将旋蒸得到的固体和100ml乙酸乙酯混合进行萃取,收集有机相,重复萃取3次,将收集的有机相合并后与无水碳酸钾混合后进行过滤,利用水泵(温度为80℃,压力为0.095bar)抽除滤液中的溶剂,得到苯乙基肼;所述苯乙基肼为有芳香气味的无色油状液体,产率为97%;
[0099]
在1℃下将6.2g 2
‑
氨基
‑
4,6
‑
二氯
‑
嘧啶
‑5‑
甲醛溶解于100ml四氢呋喃,得到第一溶液;将第一溶液和20ml三乙胺混合,得到第二溶液;将6.2g苯乙肼溶解于20ml四氢呋喃中,得到苯乙肼溶液;将苯乙肼溶液在15min内滴加至第二溶液中,25℃缩合反应24h;将缩合反应后体系旋蒸后进行层析分离,得到4
‑
氯
‑6‑
氨基
‑1‑
苯乙基吡唑并[3,4
‑
d]嘧啶;所述层析分离用层析柱为硅胶柱,层析分离用溶剂为质量比为5:1的石油醚和乙酸乙酯的混合物;所述4
‑
氯
‑6‑
氨基
‑1‑
苯乙基吡唑并[3,4
‑
d]嘧啶为浅黄色粉末状固体,产率为83%;
[0100]
将2.67g(10mmol)4
‑
氯
‑6‑
氨基
‑1‑
苯乙基吡唑并[3,4
‑
d]嘧啶,1.83g 2
‑
呋喃甲酰肼和120ml正丁醇混合80℃下取代反应12h。tcl跟踪反应,反应结束,旋蒸后在硅胶柱上进行层析(洗脱剂为质量比5:1的石油醚和乙酸乙酯的混合物),得到呋喃
‑2‑
羧酸n
’‑
(6
‑
氨基
‑1‑
苯乙基
‑
1h
‑
吡唑并[3,4
‑
d]嘧啶
‑
基)
‑4‑
酰肼;呋喃
‑2‑
羧酸n
’‑
(6
‑
氨基
‑1‑
苯乙基
‑
1h
‑
吡唑并[3,4
‑
d]嘧啶
‑
基)
‑4‑
酰肼为浅黄色泡沫状固体,产率为83%;
[0101]
将1.23g呋喃
‑2‑
羧酸n
’‑
(6
‑
氨基
‑1‑
苯乙基
‑
1h
‑
吡唑并[3,4
‑
d]嘧啶
‑
基)
‑4‑
酰肼和10ml cdi(缩合剂)混合后100℃下缩合重排反应8h,tcl跟踪反应,反应结束,旋蒸后进行层析分离(层析柱为硅胶柱,洗脱剂为质量比10:1的石油醚和乙酸乙酯的混合物),将层析分离后的产物和乙酸乙酯/石油醚混合溶剂混合后进行重结晶,得到5
‑
氨基
‑2‑
呋喃基
‑7‑
苯乙基
‑
7h
‑
吡唑并[4,3
‑
e][1,2,4]三唑并[1,5
‑
c]嘧啶;所述5
‑
氨基
‑2‑
呋喃基
‑7‑
苯乙基
‑
7h
‑
吡唑并[4,3
‑
e][1,2,4]三唑并[1,5
‑
c]嘧啶为白色粉末状固体,产率为为92%。
[0102]
实施例2
[0103]
本实施例中r1为丁基、r2为呋喃基;
[0104]
将12.6ml纯度为98%的水合肼溶解于100ml乙醇中,得到水合肼乙醇溶液;
[0105]
在回流的条件下将9g溴丁烷滴加(15min滴加完成)至水合肼乙醇溶液中,回流条件下进行58℃取代反应1.5h;将取代反应后溶液旋蒸;将旋蒸得到的固体和100ml乙酸乙酯混合进行萃取,收集有机相,重复萃取3次,将收集的有机相合并后与无水碳酸钾混合后进行过滤,利用水泵(温度为80℃,压力为0.095bar)抽除滤液中的溶剂,得到丁基肼;丁基肼,产率为90%;
[0106]
在3℃下将6.26g 2
‑
氨基
‑
4,6
‑
二氯
‑
嘧啶
‑5‑
甲醛溶解于100ml四氢呋喃,得到第一溶液;将第一溶液和20ml三乙胺混合,得到第二溶液;将6.26g丁基肼溶解于20ml四氢呋喃中,得到丁基肼溶液;将所述丁基肼溶液在15min内滴加至第二溶液中,23℃缩合反应24h;将缩合反应后体系旋蒸后进行层析分离,得到4
‑
氯
‑6‑
氨基
‑1‑
丁基吡唑并[3,4
‑
d]嘧啶;所述层析分离用层析柱为硅胶柱,所述层析分离用溶剂为质量比为5:1的石油醚和乙酸乙酯的混合物;所述4
‑
氯
‑6‑
氨基
‑1‑
丁基吡唑并[3,4
‑
d]嘧啶产率为94%;
[0107]
将2.7g(10mmol)4
‑
氯
‑6‑
氨基
‑1‑
丁基吡唑并[3,4
‑
d]嘧啶,1.86g 2
‑
呋喃甲酰肼和120ml正丁醇混合85℃下取代反应14h,tcl跟踪反应,反应结束,旋蒸后在硅胶柱上进行层析(洗脱剂为质量比5:1的石油醚和乙酸乙酯的混合物),得到呋喃
‑2‑
羧酸n
’‑
(6
‑
氨基
‑1‑
苯乙基
‑
1h
‑
吡唑并[3,4
‑
d]嘧啶
‑
基)
‑4‑
酰肼;所述呋喃
‑2‑
羧酸n
’‑
(6
‑
氨基
‑1‑
苯乙基
‑
1h
‑
吡唑并[3,4
‑
d]嘧啶
‑
基)
‑4‑
酰肼产率为86%;
[0108]
将1.25g呋喃
‑2‑
羧酸n
’‑
(6
‑
氨基
‑1‑
丁基
‑
1h
‑
吡唑并[3,4
‑
d]嘧啶
‑
基)
‑4‑
酰肼和10ml edci(缩合剂)混合后110℃下缩合重排反应10h,tcl跟踪反应,反应结束,旋蒸后进行层析分离(层析柱为硅胶柱,洗脱剂为质量比10:1的石油醚和乙酸乙酯的混合物),将层析分离后的产物和乙酸乙酯/石油醚混合溶剂混合后进行重结晶,得到5
‑
氨基
‑2‑
呋喃基
‑7‑
丁基
‑
7h
‑
吡唑并[4,3
‑
e][1,2,4]三唑并[1,5
‑
c]嘧啶;所述5
‑
氨基
‑2‑
呋喃基
‑7‑
丁基
‑
7h
‑
吡唑并[4,3
‑
e][1,2,4]三唑并[1,5
‑
c]嘧啶产率为89%。。
[0109]
实施例3
[0110]
本实施例中r1为异戊基、r2为呋喃基;
[0111]
将12.9ml纯度为98%的水合肼溶解于100ml乙醇中,得到水合肼乙醇溶液;
[0112]
在回流的条件下将9.2g异戊基溴滴加(15min滴加完成)至水合肼乙醇溶液中,回流条件下进行62℃取代反应2h;将取代反应后溶液旋蒸;将旋蒸得到的固体和100ml乙酸乙酯混合进行萃取,收集有机相,重复萃取3次,将收集的有机相合并后与无水碳酸钾混合后进行过滤,利用水泵(温度为80℃,压力为0.095bar)抽除滤液中的溶剂,得到异戊基肼;所述异戊基肼产率为94%;
[0113]
在5℃下将6.32g 2
‑
氨基
‑
4,6
‑
二氯
‑
嘧啶
‑5‑
甲醛溶解于100ml四氢呋喃,得到第一溶液;将所述第一溶液和20ml三乙胺混合,得到第二溶液;将6.32g异戊基肼溶解于20ml四氢呋喃中,得到异戊基肼溶液;将所述异戊基肼溶液在15min内滴加至第二溶液中,24℃缩合反应22h;将缩合反应后体系旋蒸后进行层析分离,得到4
‑
氯
‑6‑
氨基
‑1‑
苯乙基吡唑并[3,4
‑
d]嘧啶;所述层析分离用层析柱为硅胶柱,所述层析分离用溶剂为质量比为5:1的石油醚和乙酸乙酯的混合物;所述4
‑
氯
‑6‑
氨基
‑1‑
异戊基吡唑并[3,4
‑
d]嘧啶产率为89%;
[0114]
将2.73g(10mmol)4
‑
氯
‑6‑
氨基
‑1‑
异戊基吡唑并[3,4
‑
d]嘧啶,1.89g 2
‑
呋喃甲酰肼和120ml正丁醇混合90℃下取代反应16h,tcl跟踪反应,反应结束,旋蒸后在硅胶柱上进行层析(洗脱剂为质量比5:1的石油醚和乙酸乙酯的混合物),得到呋喃
‑2‑
羧酸n
’‑
(6
‑
氨基
‑1‑
异戊基
‑
1h
‑
吡唑并[3,4
‑
d]嘧啶
‑
基)
‑4‑
酰肼;所述呋喃
‑2‑
羧酸n
’‑
(6
‑
氨基
‑1‑
异戊基
‑
1h
‑
吡唑并[3,4
‑
d]嘧啶
‑
基)
‑4‑
酰肼产率为97%;
[0115]
将1.27g呋喃
‑2‑
羧酸n
’‑
(6
‑
氨基
‑1‑
异戊基
‑
1h
‑
吡唑并[3,4
‑
d]嘧啶
‑
基)
‑4‑
酰肼和10mltatu(缩合剂)混合后120℃下缩合重排反应12h,tcl跟踪反应,反应结束,旋蒸后进行层析分离(层析柱为硅胶柱,洗脱剂为质量比10:1的石油醚和乙酸乙酯的混合物),将层析分离后的产物和乙酸乙酯/石油醚混合溶剂混合后进行重结晶,得到5
‑
氨基
‑2‑
呋喃基
‑7‑
异戊基
‑
7h
‑
吡唑并[4,3
‑
e][1,2,4]三唑并[1,5
‑
c]嘧啶;所述5
‑
氨基
‑2‑
呋喃基
‑7‑
异戊基
‑
7h
‑
吡唑并[4,3
‑
e][1,2,4]三唑并[1,5
‑
c]嘧啶产率为为87%。
[0116]
实施例4
[0117]
本实施例中r1为苯乙基、r2为苯基;
[0118]
将13.1ml纯度为98%的水合肼溶解于100ml乙醇中,得到水合肼乙醇溶液;
[0119]
在回流的条件下将9.4g 2
‑
溴乙基苯滴加(15min滴加完成)至水合肼乙醇溶液中,回流条件下60℃进行取代反应2h;将取代反应后溶液旋蒸;将旋蒸得到的固体和100ml乙酸乙酯混合进行萃取,收集有机相,重复萃取3次,将收集的有机相合并后与无水碳酸钾混合后进行过滤,利用水泵(温度为80℃,压力为0.095bar)抽除滤液中的溶剂,得到苯乙基肼;所述苯乙基肼为有芳香气味的无色油状液体,产率为92%;
[0120]
在7℃下将6.38g 2
‑
氨基
‑
4,6
‑
二氯
‑
嘧啶
‑5‑
甲醛溶解于100ml四氢呋喃,得到第一溶液;将所述第一溶液和20ml三乙胺混合,得到第二溶液;将6.38g苯乙肼溶解于20ml四氢呋喃中,得到苯乙肼溶液;将所述苯乙肼溶液在15min内滴加至第二溶液中,25℃缩合反应24h;将缩合反应后体系旋蒸后进行层析分离,得到4
‑
氯
‑6‑
氨基
‑1‑
苯乙基吡唑并[3,4
‑
d]嘧啶;所述层析分离用层析柱为硅胶柱,所述层析分离用溶剂为质量比为5:1的石油醚和乙酸乙酯的混合物;所述4
‑
氯
‑6‑
氨基
‑1‑
苯乙基吡唑并[3,4
‑
d]嘧啶为浅黄色粉末状固体,产率为87%;
[0121]
将2.77g(10mmol)4
‑
氯
‑6‑
氨基
‑1‑
苯乙基吡唑并[3,4
‑
d]嘧啶,1.91g苯甲酰肼和120ml正丁醇混合95℃下取代反应18h,tcl跟踪反应,反应结束,旋蒸后在硅胶柱上进行层析(洗脱剂为质量比5:1的石油醚和乙酸乙酯的混合物),得到苯
‑2‑
羧酸n
’‑
(6
‑
氨基
‑1‑
苯乙基
‑
1h
‑
吡唑并[3,4
‑
d]嘧啶
‑
基)
‑4‑
酰肼;所述苯
‑2‑
羧酸n
’‑
(6
‑
氨基
‑1‑
苯乙基
‑
1h
‑
吡唑并[3,4
‑
d]嘧啶
‑
基)
‑4‑
酰肼产率为85%;
[0122]
将1.28g苯
‑2‑
羧酸n
’‑
(6
‑
氨基
‑1‑
苯乙基
‑
1h
‑
吡唑并[3,4
‑
d]嘧啶
‑
基)
‑4‑
酰肼和10ml tbtu(缩合剂)混合后130℃下缩合重排反应14h,tcl跟踪反应,反应结束后,旋蒸后进行层析分离(层析柱为硅胶柱,洗脱剂为质量比10:1的石油醚和乙酸乙酯的混合物),将层析分离后的产物和乙酸乙酯/石油醚混合溶剂混合后进行重结晶,得到5
‑
氨基
‑2‑
苯基
‑7‑
苯乙基
‑
7h
‑
吡唑并[4,3
‑
e][1,2,4]三唑并[1,5
‑
c]嘧啶;所述5
‑
氨基
‑2‑
苯基基
‑7‑
苯乙基
‑
7h
‑
吡唑并[4,3
‑
e][1,2,4]三唑并[1,5
‑
c]嘧啶产率为为85%。
[0123]
实施例5
[0124]
本实施例中r1为苯乙基,r2为吡啶基;
[0125]
将13.5ml纯度为98%的水合肼溶解于100ml乙醇中,得到水合肼乙醇溶液;
[0126]
在回流的条件下将9.6g 2
‑
溴乙基苯滴加(15min滴加完成)至水合肼乙醇溶液中,回流条件下60℃进行取代反应2h;将取代反应后溶液旋蒸;将旋蒸得到的固体和100ml乙酸乙酯混合进行萃取,收集有机相,重复萃取3次,将收集的有机相合并后与无水碳酸钾混合后进行过滤,利用水泵(温度为80℃,压力为0.095bar)抽除滤液中的溶剂,得到苯乙基肼;所述苯乙基肼为有芳香气味的无色油状液体,产率为90%;
[0127]
在9℃下将6.44g 2
‑
氨基
‑
4,6
‑
二氯
‑
嘧啶
‑5‑
甲醛溶解于100ml四氢呋喃,得到第一溶液;将所述第一溶液和20ml三乙胺混合,得到第二溶液;将6.44g苯乙肼溶解于20ml四氢呋喃中,得到苯乙肼溶液;将所述苯乙肼溶液在15min内滴加至第二溶液中,25℃缩合反应20h;将缩合反应后体系旋蒸后进行层析分离,得到4
‑
氯
‑6‑
氨基
‑1‑
苯乙基吡唑并[3,4
‑
d]嘧啶;所述层析分离用层析柱为硅胶柱,所述层析分离用溶剂为质量比为5:1的石油醚和乙酸乙酯的混合物;所述4
‑
氯
‑6‑
氨基
‑1‑
苯乙基吡唑并[3,4
‑
d]嘧啶为浅黄色粉末状固体,产率为90%;
[0128]
将2.79g(10mmol)4
‑
氯
‑6‑
氨基
‑1‑
苯乙基吡唑并[3,4
‑
d]嘧啶,1.95g 4
‑
吡啶甲酰肼和120ml正丁醇混合100℃下取代反应20h,tcl跟踪反应,反应结束,旋蒸后在硅胶柱上进行层析(洗脱剂为质量比5:1的石油醚和乙酸乙酯的混合物),得到吡啶
‑2‑
羧酸n
’‑
(6
‑
氨基
‑1‑
苯乙基
‑
1h
‑
吡唑并[3,4
‑
d]嘧啶
‑
基)
‑4‑
酰肼;所述吡啶
‑2‑
羧酸n
’‑
(6
‑
氨基
‑1‑
苯乙基
‑
1h
‑
吡唑并[3,4
‑
d]嘧啶
‑
基)
‑4‑
酰肼产率为83%;
[0129]
将1.29g吡啶
‑2‑
羧酸n
’‑
(6
‑
氨基
‑1‑
苯乙基
‑
1h
‑
吡唑并[3,4
‑
d]嘧啶
‑
基)
‑4‑
酰肼和10mltatu(缩合剂)混合后140℃下缩合重排反应16h,tcl跟踪反应,反应结束,旋蒸后进行层析分离(层析柱为硅胶柱,洗脱剂为质量比10:1的石油醚和乙酸乙酯的混合物),将层析分离后的产物和乙酸乙酯/石油醚混合溶剂混合后进行重结晶,得到5
‑
氨基
‑4‑
吡啶基
‑7‑
苯乙基
‑
7h
‑
吡唑并[4,3
‑
e][1,2,4]三唑并[1,5
‑
c]嘧啶;所述5
‑
氨基
‑4‑
吡啶基
‑7‑
苯乙基
‑
7h
‑
吡唑并[4,3
‑
e][1,2,4]三唑并[1,5
‑
c]嘧啶产率为93%。
[0130]
将实施例1制备得到的5
‑
氨基
‑2‑
呋喃基
‑7‑
苯乙基
‑
7h
‑
吡唑并[4,3
‑
e][1,2,4]三唑并[1,5
‑
c]嘧啶进行核磁检测,得到核磁谱图如图1~2所示。其中图1为实施例1制备得到的5
‑
氨基
‑2‑
呋喃基
‑7‑
苯乙基
‑
7h
‑
吡唑并[4,3
‑
e][1,2,4]三唑并[1,5
‑
c]嘧啶的核磁共振氢谱图,图2为实施例1制备得到的5
‑
氨基
‑2‑
呋喃基
‑7‑
苯乙基
‑
7h
‑
吡唑并[4,3
‑
e][1,2,4]三唑并[1,5
‑
c]嘧啶的核磁共振碳谱图。
[0131]
由图1和图2可知,按照本发明制备方法能够制备得到吡唑并嘧啶并三唑环类化合物。
[0132]
尽管上述实施例对本发明做出了详尽的描述,但它仅仅是本发明一部分实施例,而不是全部实施例,人们还可以根据本实施例在不经创造性前提下获得其他实施例,这些实施例都属于本发明保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1