一种金属离子萃取剂及其合成方法与流程
1.本发明涉及萃取剂的合成技术领域,特别是涉及一种金属离子萃取剂及其合成方法。
背景技术:
2.稀土是重要的有色金属,在新材料开发、超纯材料制备等领域具有巨大的应用价值。溶剂萃取具有分离效率高、能耗低的特点,已成为分离纯化稀土的重要手段。另外,核燃料后处理时,利用中子将半衰期很长的锕系元素转变为半衰期短的同位素,但镧系元素能有效捕获中子,所以必须利用溶剂萃取的方法将高放废液中的镧系元素和锕系元素分离。新型萃取剂的研制和科学设计工艺流程一直是溶剂萃取的两个重要方面,因此探索高效、廉价的萃取剂对萃取化学的发展具有重大意义。
3.含硫萃取剂虽然广泛用于提取部分重金属、镧系元素及贵金属。但缺点是pka值较高,适宜萃取的体系酸度过低,且容易酸解,在一定程度上限制了应用;以及焚烧后会造成二次污染。酰胺萃取剂具有易合成、耐辐射和能完全燃尽无污染等优点,是新型绿色环保萃取剂。但缺点是取代酰胺失去了氢键缔合能力,其水溶性较差。且氮原子未被完全取代,该萃取剂未能充分发挥配位效应,萃取能力一般。双酰胺荚醚类萃取剂是近年来提出的一类新型绿色萃取剂,相比于酰胺萃取剂,该类萃取剂以三齿配位方式与金属离子络合,提高了萃合物的稳定性,从而提高萃取效率,如n,n,n’,n
’‑
四辛基
‑3‑
氧戊二酰胺(todga)萃取剂,但该萃取剂在酸度过大或金属离子浓度过高的体系中易出现相分离的现象。而如何制备得到一种与金属的结合效果好、选择性好、在酸度过大或金属离子浓度过高的体系不易出现相分离的高效的、绿色的萃取剂成为本领域技术人员亟待解决的技术难题。
技术实现要素:
4.本发明的目的是提供一种金属离子萃取剂及其合成方法,以解决上述现有技术存在的问题,通过改变原料及制备方法,制备得到了一种碳链长度相对较短,空间位阻小,与金属结合效果好、选择性好的高效、绿色的萃取剂。
5.为实现上述目的,本发明提供了如下方案:
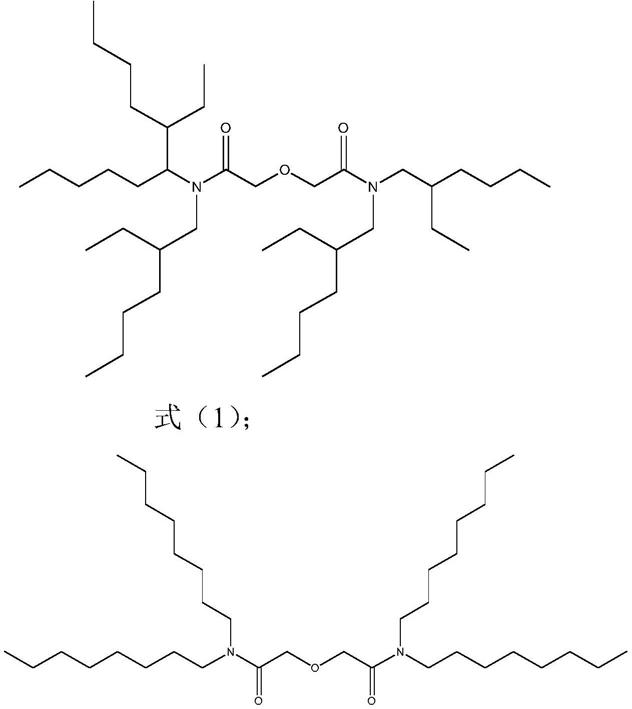
6.本发明的技术方案之一:一种金属离子萃取剂,所述金属离子萃取剂的结构式如式(1)或式(2)所示:
[0007][0008][0009]
本发明的技术方案之二:一种根据权利要求1所述的金属离子萃取剂的合成方法,所述式(1)所示萃取剂的合成方法包括以下步骤:
[0010]
(1)在二甘醇中加入无机酸加热回流反应得到二甘醇酸;
[0011]
(2)在步骤(1)中制备得到的二甘醇酸中加入亚硫酰氯反应得到二甘醇酰氯;
[0012]
(3)在2
‑
乙基己胺和无机碱的混合溶液中加入步骤(2)中制备得到的二甘醇酰氯反应得到式(1)所示萃取剂。
[0013]
本发明的技术方案之三:一种根据权利要求1所述的金属离子萃取剂的合成方法,所述式(2)所示萃取剂的合成方法包括以下步骤:
[0014]
(1)在二甘醇中加入无机酸加热回流反应得到二甘醇酸;
[0015]
(2)在步骤(1)中制备得到的二甘醇酸中加入亚硫酰氯加热回流反应得到二甘醇酰氯;
[0016]
(3)在二正辛胺和无机碱的混合溶液中加入步骤(2)中制备得到的二甘醇酰氯反应得到式(2)所示萃取剂。
[0017]
进一步地,所述步骤(1)具体包括:在二甘醇中加入无机酸一次加热反应至无气体释放后,再加入二甘醇二次加热反应得到二甘醇酸。
[0018]
进一步地,所述无机酸在二甘醇加热至45℃时加入;所述无机酸为硝酸;所述无机酸的摩尔浓度为0.01mol/l;所述一次加热反应和二次加热反应中的二甘醇的摩尔比为1:1~1:6;所述一次加热反应和二次加热反应中的二甘醇总的摩尔质量与无机酸的摩尔质量
比为1:0.001~0.004。
[0019]
进一步地,所述一次加热的温度为60~70℃;所述二次加热具体包括:40~50℃下保持30~60min,然后升温至75~85℃下加热反应20~40min。
[0020]
进一步地,所述步骤(2)中二甘醇酸与亚硫酰氯的摩尔比为1:3~1:8,加热回流的时间为4~8h。
[0021]
进一步地,所述步骤(3)中二甘醇酰氯与2
‑
乙基己胺的摩尔质量比为1:1~1:6;所述2
‑
乙基己胺与无机碱的摩尔质量比为1:20~1:50。
[0022]
进一步地,所述步骤(3)中二甘醇酰氯与二正辛胺的摩尔质量比为1:0.5~1:5;所述二正辛胺与无机碱的摩尔质量比为1:60~1:80。
[0023]
进一步地,所述步骤(3)中的无机碱为氢氧化钠;所述无机碱的质量分数为30%。
[0024]
本发明公开了以下技术效果:
[0025]
本发明制备得到的金属离子萃取剂克服了现有萃取剂不够绿色环保、萃取效果一般及酸度大体系中易形成多相分离的缺点,具有碳链长度相对较短,空间位阻小等优点,且与金属结合效果好、选择性好,萃取效率高,是一种绿色的萃取剂。
附图说明
[0026]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0027]
图1为本发明实施例1的合成路线图,1为硝酸,2为亚硫酰氯,3为2
‑
乙基己胺;
[0028]
图2为本发明实施例1制备得到的n,n,n’,n
’‑
四
‑2‑
乙基己基二甘醇酰胺的1hnmr光谱图;
[0029]
图3为本发明实施例2制备得到的n,n,n’,n
’‑
四辛基二甘醇酰胺的1hnmr光谱图。
具体实施方式
[0030]
现详细说明本发明的多种示例性实施方式,该详细说明不应认为是对本发明的限制,而应理解为是对本发明的某些方面、特性和实施方案的更详细的描述。
[0031]
应理解本发明中所述的术语仅仅是为描述特别的实施方式,并非用于限制本发明。另外,对于本发明中的数值范围,应理解为还具体公开了该范围的上限和下限之间的每个中间值。在任何陈述值或陈述范围内的中间值以及任何其他陈述值或在所述范围内的中间值之间的每个较小的范围也包括在本发明内。这些较小范围的上限和下限可独立地包括或排除在范围内。
[0032]
除非另有说明,否则本文使用的所有技术和科学术语具有本发明所述领域的常规技术人员通常理解的相同含义。虽然本发明仅描述了优选的方法和材料,但是在本发明的实施或测试中也可以使用与本文所述相似或等同的任何方法和材料。本说明书中提到的所有文献通过引用并入,用以公开和描述与所述文献相关的方法和/或材料。在与任何并入的文献冲突时,以本说明书的内容为准。
[0033]
在不背离本发明的范围或精神的情况下,可对本发明说明书的具体实施方式做多
种改进和变化,这对本领域技术人员而言是显而易见的。由本发明的说明书得到的其他实施方式对技术人员而言是显而易见的。本技术说明书和实施例仅是示例性的。
[0034]
关于本文中所使用的“包含”、“包括”、“具有”、“含有”等等,均为开放性的用语,即意指包含但不限于。
[0035]
实施例1
[0036]
n,n,n’,n
’‑
四
‑2‑
乙基己基二甘醇酰胺的合成方法:
[0037]
(1)在洁净的烧杯中加入1.0g(0.0094mol)二甘醇,升温至45℃后加入15ml摩尔浓度为0.01mol/l的硝酸,然后加热至65℃反应至无氮氧化物(no2)释放结束,冷却至45~50℃后,在缓慢加入4.9g(0.0462mol)二甘醇,加入时间为1h;二甘醇加样完成后继续在45℃下保持40min,然后升温至80℃反应30min,反应结束后在70℃真空下除去溶剂,用迪安
‑
斯达克分水器与苯共沸蒸馏干燥残留物,然后将产物过滤并从丙酮
‑
苯混合物中重结晶得到二甘醇酸,产率为92%。
[0038]
(2)在干燥烧瓶中25g(0.187mol)二甘醇酸,然后加入85ml(约139.23g,1.17mol)亚硫酰氯后立即加热至回流并在该温度下搅拌反应5h,、反应结束后,在85℃下减压蒸馏除去过量的亚硫酰氯,然后在室温下高真空直至混合物沉淀,过滤得到沉淀物,将沉淀物在40℃下高真空和升高温度下蒸馏,直到收集到所有二甘醇酰氯液体,产率为95%。
[0039]
(3)在干燥烧瓶中加入0.83g(0.0064mol)2
‑
乙基己胺和30ml 30%的naoh溶液冷却至0℃,保持温度不变,逐滴滴加二甘醇酰氯的乙醚溶液(二甘醇酰氯的乙醚溶液中含有0.85g(0.005mol)二甘醇酰氯)滴加时间为30min,滴加完成后0℃条件下搅拌反应2h,静置分层,在水相中加入饱和nacl溶液清洗后用乙醚萃取回收未反应的二甘醇酰氯;在有机相中加入20ml 10%hcl水溶液,剧烈摇动促进铵盐团块的形成,并用10%hcl水溶液洗涤两次铵盐团块,然后用玻璃料(g3)过滤得到滤液,在滤液中加入25ml质量分数为4%的聚(4
‑
苯乙烯磺酸)的水溶液,剧烈摇动10min(以促进聚合物盐作为漂浮在层之间的无定形材料的形成),然后用蒸馏水洗涤去除为反应的聚(4
‑
苯乙烯磺酸),得到的有机溶液用mgso4干燥,然后通过真空蒸发除去溶剂,得到n,n,n’,n
’‑
四
‑2‑
乙基己基二甘醇酰胺(tedga),根据1hnmr光谱(见图2)得知其纯度≥97%,产率为94%。
[0040]
反应流程图见图1;图中1为硝酸,2为亚硫酰氯,3为2
‑
乙基己胺。
[0041]
实施例2
[0042]
n,n,n’,n
’‑
四辛基二甘醇酰胺的合成方法:
[0043]
同实施例1,区别在于,将步骤(3)中的2
‑
乙基己胺替换成0.83g(0.0034mol)二正辛胺,制备得到n,n,n’,n
’‑
四辛基二甘醇酰胺(todga),根据1hnmr光谱(见图3)得知其纯度≥97%,产率为91%。
[0044]
实施例3
[0045]
n,n,n’,n
’‑
四
‑2‑
乙基己基二甘醇酰胺的合成方法:
[0046]
同实施例1,区别在于,步骤(1)具体为在洁净的烧杯中加入1.0g(0.0094mol)二甘醇,升温至45℃后加入15ml摩尔浓度为0.1mol/l的硝酸,然后加热至60℃反应至无氮氧化物(no2)释放结束,冷却至45~50℃后,在缓慢加入4.9g(0.0462mol)二甘醇,加入时间为1h;二甘醇加样完成后继续在40℃下保持40min,然后升温至85℃反应20min,反应结束后在70℃真空下除去溶剂,用迪安
‑
斯达克分水器与苯共沸蒸馏干燥残留物,然后将产物过滤并
从丙酮
‑
苯混合物中重结晶得到二甘酸,产率为73%。
[0047]
实施例4
[0048]
n,n,n’,n
’‑
四
‑2‑
乙基己基二甘醇酰胺的合成方法:
[0049]
同实施例1,区别在于,步骤(1)中硝酸的加入量为10ml,产率为65%。
[0050]
实施例5
[0051]
n,n,n’,n
’‑
四
‑2‑
乙基己基二甘醇酰胺的合成方法:
[0052]
同实施例1,区别在于,步骤(2)中亚硫酰氯的加入量为60ml,产率为60%。
[0053]
对比例1
[0054]
n,n,n’,n
’‑
四
‑2‑
乙基己基二甘醇酰胺的合成方法:
[0055]
同实施例1,区别在于,步骤(1)具体为在洁净的烧杯中加入15ml摩尔浓度为0.01mol/l的硝酸,升温至45~50℃后,再缓慢加入5.9g(0.0556mol)二甘醇,加入时间为1h;二甘醇加样完成后继续在40℃下保持40min,然后升温至85℃反应20min,反应结束后在70℃真空下除去溶剂,用迪安
‑
斯达克分水器与苯共沸蒸馏干燥残留物,然后将产物过滤并从丙酮
‑
苯混合物中重结晶得到二甘酸,产率为70%。
[0056]
以上所述的实施例仅是对本发明的优选方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案做出的各种变形和改进,均应落入本发明权利要求书确定的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1