一种提高CHO细胞同源重组效率的方法及其相关产品和应用
一种提高cho细胞同源重组效率的方法及其相关产品和应用
技术领域
1.本发明属于生物医药技术领域,具体而言,本发明涉及一种提高同源重组效率的方法,更具体地,本发明涉及一种提高cho细胞同源重组效率的方法及其相关产品和应用。
背景技术:
2.中国仓鼠卵巢细胞(chinese hamster ovary cells,cho)是工业生产重组蛋白类生物药物最主要的细胞株,自1957年dr.theodore t.puck从成年雌性仓鼠卵巢分离获得第一株野生型cho细胞以来,经过多次改造逐渐形成以dhfr
‑
cho(缺失二氢叶酸还原酶)表达系统和gs
‑
cho(缺失谷氨酰胺合成酶)表达系统为主的商业化表达系统。目前,cho细胞已经是用于包括基因工程抗体类药物在内的重组糖蛋白药物生产的首选宿主细胞,主要有以下原因:1、cho细胞能够较好地适应悬浮培养,在生物反应器中可获得较高的密度,有利于工业上大规模培养;2、cho细胞能够在翻译后进行修饰,尤其是在蛋白糖基化方面,与人类基本一致;3、利于外源基因整合,具有外源重组基因的高效扩增和表达能力;4、cho细胞属于成纤维细胞,是一种非分泌型细胞,很少分泌自身的内源蛋白,因此十分有利于下游目标蛋白的分离纯化;5、cho细胞能够在无血清和化学成分明确的培养基中培养,能够确保不同批次培养的重复性;6、cho细胞不易传播人类感染病毒。目前,构建工业化稳转重组细胞株的方法主要是通过随机转基因技术或者有专利保护的商业化的位点特异重组酶为基础的flip
‑
in技术进行制备,存在外源基因重组效率不高以及筛选高表达工程细胞株耗时耗力等方面的问题。
3.以同源重组为基础的基因打靶技术(gene targeting)的出现,能定点的敲入外源基因(gene insertion),解除转基因技术随机插入的问题,同时结合rosa26、h11、hprt、c12orf35等安全位点技术,可以将外源基因定向的整合到安全位点,这样可以保证外源基因持续稳定的高表达。然而传统的基因打靶效率很低、目的基因大概率进行随机整合而非定点敲入,而且通常在小鼠的胚胎干细胞(es细胞)中完成。在大型哺乳动物原代及其他类型细胞的基因打靶效率更低,大大限制了其应用。直到第一代锌指核酸酶(zinc
‑
finger nucleaaes,zfns)基因编辑技术,以及随后的第二代转录激活因子样效应物核酸酶(transcription activator
‑
like effector nucleases,talens)、第三代规律成簇间隔短回文重复(clustered regularly interspaced short palindromic repeats,crispr)/cas9技术的出现,才提高了此技术的效率及应用面。2010年orlando sj等研究人员首次利用zfns技术以寡核苷酸(oligonucleotide)或者短的供体载体为模板在cho细胞的内源gs基因进行非同源重组依赖的基因敲入。随后cristea于2013年首次利用zfns和talens技术结合体内双切供体载体,完成cho细胞上的非同源臂依赖的基因敲入。虽然目的基因可以整合到目的基因区域,但并不是精确的同源重组,因为即使有基因编辑技术,cho细胞自身同源重组效率依然很低。2017年kawabe y研究团队利用crispr/cas9和mmej的pitch技术成功对cho细胞进行基因敲入,同样也是非同源重组依赖的。2016年jae seong lee团队首次利用crispr/cas9、同源重组技术及荧光报告系统富集技术在cho细胞中完成了目的基因的定
点敲入。虽然少数研究证明cho细胞可以实现目的基因精确的定点敲入,但总体的同源重组效率远远低于目的基因随机整合效率。因此,提高cho细胞同源重组效率成为了目前本领域亟待解决的技术问题。
4.传统的转基因技术具有外源基因随机的,多拷贝插入等缺陷,这样通常造成位置效应和基因沉默,使外源基因不能持续稳定的表达。同时由于多拷贝的插入,很难确定外源基因具体的插入位置,也容易造成受体细胞基因组的插入突变,对受体细胞本身造成潜在危害。这些都大大限制了转基因技术制备cho细胞生物反应器的发展和应用。随后国外公司利用逆转录病毒随机筛选法,获得了可以高效支持外源基因表达的位点,并且建立flip
‑
in技术制备了商业化细胞系,通过位点重组酶系统进行外源基因的定点敲入,但此技术的位点属于商业机密,未对外公开,相关定点cho表达系统只能通过商业化购买,价格昂贵,且操作繁琐,具有很强的技术壁垒,大大限制cho细胞生物反应器的通用性和实用性。因此解决上述技术缺陷对于构建cho宿主细胞而言十分重要。
5.最近,相关研究学者通过实验研究证明了在小鼠胚胎干细胞及人b淋巴细胞白血病细胞(nalm6细胞)中对lig4基因和polq基因同时进行敲除,从而抑制宿主细胞的nhej修复途径进而抑制随机整合,提高细胞的同源整合效率,但是在该研究中单独敲除polq基因,并没有明显提高同源重组效率。以往诸多研究表明cho细胞是一个同源重组效率很低的细胞系,目前在该细胞中导入外源基因表达都是通过随机整合和重组酶介导的位点特异重组技术来介导。本发明首次在cho细胞中证明单独敲除polq基因,抑制al
‑
nhej修复途径可以高效提高cho细胞同源重组效率,对于其在细胞生物反应器中意义重大。本发明首次利用crispr/cas9技术成功制备了polq基因敲除的cho
‑
k1工具细胞系,并验证此工具细胞系发生同源重组的效率远高于野生型的cho
‑
k1细胞系。此技术为cho细胞高效的精确整合目的基因及高效表达外源目的蛋白提供了有效的途径。
技术实现要素:
6.为了解决当前本领域现有技术存在的上述技术缺陷,本发明的目的在于提供一种提高cho细胞同源重组效率的方法及其相关产品和应用。
7.本发明的上述目的通过以下技术方案得以实现:
8.本发明的第一方面提供了一种提高cho细胞同源重组效率的方法。
9.进一步,所述方法包括敲除cho细胞中的polq基因和/或抑制cho细胞中polq基因的表达。
10.进一步,所述cho细胞为中国仓鼠卵巢细胞(chinese hamster ovary cells,cho),是工业生产重组蛋白类生物药物最主要的细胞株,cho细胞目前已经成为用于包括基因工程抗体类药物在内的重组糖蛋白药物生产的首选宿主细胞;
11.优选地,所述cho细胞为cho
‑
k1细胞。
12.进一步,所述方法包括如下步骤:
13.步骤(1):利用基因编辑技术将cho细胞中的polq基因敲除和/或失活;
14.步骤(2):细胞分选,对经细胞分选得到的polq基因敲除和/或失活的cho细胞进行鉴定和筛选;
15.优选地,步骤(1)中所述基因编辑技术包括crispr/cas9、cas12a、spry
‑
cas9、spg
‑
cas9及相关突变体、zfns、talens;
16.更优选地,步骤(1)中所述基因编辑技术为crispr/cas9。
17.进一步,步骤(1)中所述的利用crispr/cas9基因编辑技术将cho细胞中的polq基因敲除包括如下步骤:
18.步骤(a):针对仓鼠polq基因设计靶向polq基因的sgrna的靶向识别区域;
19.步骤(b):将步骤(a)得到的成对的sgrna退火、配对,获得带有粘性末端的双链dna片段;
20.步骤(c):将步骤(b)得到的双链dna片段与酶切后的cas9载体连接,获得重组表达载体;
21.步骤(d):将步骤(c)得到的重组表达载体转染cho细胞,培养,获得polq基因敲除的cho细胞;
22.优选地,步骤(a)中所述的sgrna的靶向识别区域的序列如seq id no:2所示;
23.优选地,步骤(b)中所述的退火的条件为100℃、5min;
24.优选地,步骤(c)中所述的cas9及相关突变体载体包括px330载体、px460载体、px459载体、px458载体、px552载体、px551载体、px856载体、px855载体、px854载体、px853载体、px852载体、px851载体、px603载体、px602载体、px601载体、px600载体、px399载体、px398载体、px396载体、px395载体、px393载体、px389载体、px388载体、px387载体、px386载体、px335载体、px334载体、px260载体、px165载体;
25.更优选地,步骤(c)中所述的cas9载体为px330载体;
26.最优选地,步骤(c)中所述的酶切后的cas9载体为bbsi限制性核酸内切酶特异性切割后得到的特异线性质粒;
27.优选地,步骤(d)中所述的转染的过程包括将步骤(c)得到的重组表达载体与绿色荧光蛋白表达质粒共转染cho细胞;
28.更优选地,所述绿色荧光蛋白表达质粒为pmax
‑
gfp。
29.进一步,所述cho细胞选自cho
‑
k1、cho
‑
s、cho
‑
dg44、cho
‑
dxb11中的任意一种;
30.优选地,在本发明的具体实施方案中所述cho细胞为cho
‑
k1。
31.进一步,所述方法中还包括对重组表达载体的基因编辑效率进行验证,验证的方法包括如下步骤:流式分选富集gfp阳性细胞后用细胞基因组提取试剂盒提取细胞基因组,扩增polq基因,pcr扩增产物柱纯化回收后进行梯度退火,用t7ei酶对退火产物进行酶切后,琼脂糖凝胶电泳检测切割条带,并根据切割带的灰度值验证载体编辑效率。
32.进一步,所述扩增polq基因的pcr上游引物的序列如seq id no:5所示,所述扩增polq基因的pcr下游引物的序列如seq id no:6所示。
33.本发明通过上述方法获得了敲除polq基因的高效定点整合外源基因的cho工程细胞株,利用该细胞株能够高效定点整合外源基因,显著提高外源基因的整合效率,进而高效表达目的蛋白,克服了目前构建稳转cho细胞株时存在的筛选周期长、插入位点随机性、易产生位置效应和基因沉默等技术问题,此外,构建所述cho工程细胞株中未引入新的筛选标记基因,最大限度地保证了原宿主基因组完整性。
34.进一步,本发明所述提高cho细胞同源重组效率的方法还包括同时敲除cho细胞中的polq基因和lig4基因和/或同时抑制cho细胞中polq基因和lig4基因的表达;
35.优选地,所述提高cho细胞同源重组效率的方法还包括同时敲除cho细胞中的polq基因和lig4基因;
36.更优选地,所述同时敲除cho细胞中的polq基因和lig4基因的方法包括如下步骤:在上述构建好的polq敲除细胞株基础上针对lig4进行敲除并进行鉴定;
37.最优选地,所述针对lig4基因的靶点序列如seq id no:7所示。
38.本发明的第二方面提供了一种用于提高cho细胞同源重组效率的靶向polq基因的sgrna。
39.进一步,所述sgrna的序列如seq id no:2所示。
40.本发明的第三方面提供了一种用于提高cho细胞同源重组效率的crispr/cas9基因编辑系统。
41.进一步,所述基因编辑系统包含cas9和本发明第二方面所述的sgrna。
42.本发明的第四方面提供了一种高效定点整合外源基因的cho工程细胞株。
43.进一步,所述cho工程细胞株为敲除cho细胞中的polq基因和/或抑制cho细胞中polq基因的表达得到的细胞株;
44.优选地,所述cho工程细胞株为根据本发明第一方面所述的方法制备得到的细胞株。
45.进一步,利用上述cho工程细胞株能够高效定点精准整合外源基因,显著提高外源基因的整合效率,进而高效表达目的蛋白。
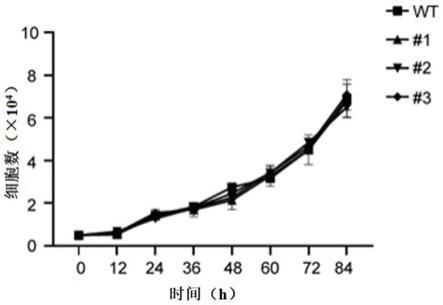
46.本发明的第五方面提供了一种使外源基因在cho细胞中持续高表达的方法。
47.进一步,所述方法包括将外源基因插入到本发明第四方面所述的cho工程细胞株基因组的安全位点进行同源重组;
48.优选地,所述安全位点包括rosa26、h11、hprt、c12orf35、aavs1、ccr5、col1a1、tigre;
49.更优选地,所述安全位点为rosa26。
50.本发明的第六方面提供了一种cho细胞中rosa26特异性整合位点的同源重组供体载体。
51.进一步,所述同源重组供体载体包含如下元件:cho细胞rosa26 1kb 5’同源臂序列和1kb 3’同源臂序列、sa内含子剪切信号、无启动子的egfp基因。
52.进一步,在本发明构建得到的敲除polq基因的cho工程细胞株中,只有发生同源重组时,细胞才能变绿(细胞发绿色荧光),因此,简单的通过流式分选即可统计同源重组的效率,可快速实现同源重组效率的检测。
53.本发明的第七方面提供了一种快速检测cho细胞同源重组效率的方法。
54.进一步,所述方法包括利用crispr/cas9基因编辑技术将本发明第六方面所述的同源重组供体载体和靶向安全位点的sgrna定点敲入到cho细胞的安全位点,然后通过流式细胞分选的方法统计荧光蛋白的表达来反应同源重组的效率;
55.优选地,所述安全位点包括rosa26、h11、hprt、c12orf35、aavs1、ccr5、col1a1、tigre;
56.更优选地,所述安全位点为rosa26;
57.最优选地,所述靶向安全位点的sgrna的序列如seq id no:4所示。
58.本发明的第八方面提供了如下任一方面的应用:
59.(1)本发明第二方面所述的sgrna在提高cho细胞同源重组效率中的应用;
60.(2)本发明第三方面所述的基因编辑系统在提高cho细胞同源重组效率中的应用;
61.(3)用于敲除polq基因的试剂和/或抑制polq基因表达水平的试剂在提高cho细胞同源重组效率中的应用;
62.(4)本发明第二方面所述的sgrna在制备高效定点整合外源基因的cho工程细胞株中的应用;
63.(5)本发明第三方面所述的基因编辑系统在制备高效定点整合外源基因的cho工程细胞株中的应用;
64.(6)用于敲除polq基因的试剂和/或抑制polq基因表达水平的试剂在制备高效定点整合外源基因的cho工程细胞株中的应用;
65.(7)本发明第四方面所述的cho工程细胞株在构建重组蛋白表达系统中的应用;
66.(8)本发明第四方面所述的cho工程细胞株在制备重组蛋白类生物药物中的应用;
67.(9)本发明第六方面所述的同源重组供体载体在快速检测cho细胞同源重组效率中的应用。
68.进一步,所述重组蛋白表达系统由包括如下步骤的方法制备得到:将目的基因插入表达载体中,构建得到重组蛋白表达载体;将该重组蛋白表达载体转染至如本发明第四方面所述的cho工程细胞株中,经筛选得到重组cho细胞表达系统,所述重组cho细胞表达系统能够高效表达目的蛋白。
69.进一步,所述cho工程细胞株在制备重组蛋白类生物药物中的应用包括如下步骤:将感兴趣的目的基因插入表达载体中,构建得到重组蛋白表达载体;将该重组蛋白表达载体转染至如本发明第四方面所述的cho工程细胞株中,经筛选得到重组cho细胞表达系统,所述重组cho细胞表达系统即可高效表达感兴趣的目的蛋白,所述目的蛋白即为重组蛋白类生物药物的有效成分。
70.为了进一步解释本发明,对本发明中涉及到的专业科学术语作如下解释:
71.本文中使用的术语“sgrna”,即为小向导rna(small guide rna,sgrna)、向导rna或引导rna(guide rna,grna),作用于动质体(kinetoplastid)体内一种称为rna编辑(rna editing)的后转录修饰过程中,也是一种小型非编码rna。可与pre
‑
mrna配对,并在其中插入一些尿嘧啶(u),产生具有作用的mrna。向导rna编辑的rna分子,长度大约是60
‑
80个核苷酸,是由单独的基因转录的,在grna的5’末端有一段锚定区,以特殊的g
‑
u配对方式与非编辑的pre
‑
mrna序列互补,锚定序列促进grna与pre
‑
mrna中的编辑区互补,特意结合;在grna分子的中间部位有一个编辑区负责在被编辑的pre
‑
mrna分子中插入u的位置,其与被编辑mrna精确互补;在grna分子的3’末端,有一段转录后加入的由大约15个非编码的polyu序列,功能为把grna链接到pre
‑
mrna的编辑区的5’上游富含嘌呤碱基的核苷酸序列上。在编辑时,形成一个编辑体(editosome),以grna内部的序列作为模板进行转录物的校正,同时产生编辑的mrna。
72.本文中使用的术语“同源重组(homologous recombination,hr)”,是指发生在非姐妹染色单体之间或同一染色体上含有同源序列的dna分子之间或分子之内的重新组合。同源重组需要一系列的蛋白质催化,如原核生物细胞内的reca、recbcd、recf、reco、recr
等;以及真核生物细胞内的rad51、mre11
‑
rad50等。同源重组反应通常根据交叉分子或holliday结构的形成和拆分分为三个阶段,即前联会体阶段、联会体形成和holliday结构的拆分。同源重组反应依赖于dna分子之间的同源性,100%同源性的dna分子之间的重组常见于非姐妹染色体之间的同源重组,称为homologous recombination,而小于100%同源性的dna分子之间或分子之内的重组,则被称为hemologus recombination。后者可被负责碱基错配对的蛋白如原核细胞内的muts或真核生物细胞内的msh2
‑
3等蛋白质“编辑”。同源重组可以双向交换dna分子,也可以单向转移dna分子,后者又被称为基因转换(gene conversion)。
73.本文中使用的术语“野生型(wild type,wt)”,是指生物、菌株、基因的典型形式或者当它在自然界存在时区别于突变体或变体形式的特征。
74.本文中使用的术语“突变体(mutant)”,是指发生突变的个体,其具有与野生型不同的序列,可能会导致其中序列的至少部分功能已丢失的序列,例如,在启动子或增强子区域中序列的变化将至少部分地影响生物体中编码序列的表达。术语“突变”是指可由诸如缺失、添加、取代或重排引起的核酸序列中序列的任何变化。突变还可影响该序列参与的一个或多个步骤。例如,dna序列中的变化可导致有活性的、有部分活性的或无活性的改变的mrna和/或蛋白的合成。
75.本文中使用的术语“表达”,是指感兴趣的序列转录产生对应mrna并且该mrna翻译产生对应产物,即肽、多肽或蛋白。调节元件控制或调整感兴趣的序列表达,所述调节元件包括5’调节元件如启动子。
76.本文中使用的术语“重组表达载体”,是指来自任何来源、能够整合入基因组或自主复制的任何因子如质粒、粘粒、病毒、bac(细菌人工染色体)、自主复制型序列、噬菌体、或者线性或环状单链或双链dna或rna核苷酸序列,包括其中一种或多种dna序列使用众所周知的重组dna技术以功能性可操作方式连接的dna分子。
77.本文中使用的术语“引物”,是指一段分离的核酸分子,其通过核酸杂交,退火结合到互补的目标dna链上,在引物和目标dna链之间形成杂合体,然后在聚合酶(例如dna聚合酶)的作用下,沿目标dna链延伸。本发明的引物对涉及其在目标核酸序列扩增中的应用,例如,通过聚合酶链式反应(pcr)或其他常规的核酸扩增方法。
78.相对于现有技术,本发明具有的优点和有益效果:
79.(1)本发明首次在cho细胞中证明单独敲除polq基因,抑制al
‑
nhej修复途径可以高效提高cho细胞同源重组效率,对于其在细胞生物反应器中意义重大,构建得到的polq基因敲除的cho工具细胞系能够提高今后表达外源抗体时筛选稳定转染细胞株的效率,有利于进一步提高产物的表达量;
80.(2)本发明首次利用crispr/cas9技术成功制备了polq基因敲除的cho工具细胞系,并验证此工具细胞系发生同源重组的效率远高于野生型的cho细胞系;该技术为cho细胞高效的精确整合目的基因及高效表达外源目的蛋白提供了有效的途径;
81.(3)以往的研究不论是在肿瘤细胞还是在es细胞中,敲除polq基因,均会导致细胞的生长抑制,在动物中敲除polq会导致动物异常甚至致死,然而本发明所述的利用crispr/cas9技术敲除polq基因得到的敲除polq
‑
/
‑
cho细胞的生长未受到影响,且状态良好。
附图说明
82.以下,结合附图来详细说明本发明的实施方案,其中:
83.图1显示cho细胞中针对polq的靶点序列及crispr/cas9载体示意图;
84.图2显示靶向polq基因sgrna序列图;
85.图3显示t7ei及ta克隆验证针对polq基因的crispr/cas9载体效率的结果图;
86.图4显示cho细胞中针对rosa26基因的靶点序列及同源重组效率验证载体示意图;
87.图5显示三株经polq基因敲除、lig4基因敲除、polq基因和lig4基因双敲除的cho单克隆细胞株的鉴定结果图,其中,a图:polq基因敲除的细胞株的测序结果图,b图:polq基因敲除的细胞株电泳结果图,c图:lig4基因敲除的细胞株的测序结果图,d图:polq基因和lig4基因双敲除的细胞株的测序结果图;
88.图6显示流式分析统计同源重组效率的结果图,其中,a图:流式分析结果图,b图:流式分析结果统计图;
89.图7显示野生型cho
‑
k1细胞及本发明构建得到的敲除polq基因的cho
‑
k1细胞突变株的生长曲线图。
具体实施方式
90.下面结合具体实施例,进一步阐述本发明,仅用于解释本发明,而不能理解为对本发明的限制。本领域的普通技术人员可以理解为:在不脱离本发明的原理和宗旨的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由权利要求及其等同物限定。下列实施例中未注明具体条件的实验方法,通常按照常规条件或按照厂商所建议的条件实施检测。
91.实施例1构建polq基因的crispr/cas9载体及其基因编辑的效率验证
92.1、实验材料
93.cho
‑
k1(ccl
‑
61
tm
)细胞购自atcc公司;dmem f12培养基、磷酸缓冲液、0.25%
‑
edta胰酶购自gibico公司;胎牛血清购自四季青公司;px330、ploxp2neo购自addgene公司;polq基因扩增引物、单链向导rna(sgrna)寡核苷酸及rosa26
‑
sa
‑
egfp
‑
polya的dna序列合成及基因测序由上海生工生物有限公司完成;t4 dna连接酶、2xq5 mix预混试剂、t7ei酶、核酸内切酶bbsi、noti、xhoi及sali购自neb公司;细胞基因组dna提取试剂盒购自天根生化公司;氯仿、异丙醇、无水乙醇、生理盐水购自国药集团化学试剂有限公司;无内毒素质粒大提试剂盒及pcr产物纯化试剂盒购自qiagen公司;tranzol up购自康为世纪;逆转录试剂盒购自东洋纺(toyobo)公司;top10感受态细胞购自博迈德生物;pmax
‑
gfp质粒购自lonza公司;细胞转染试剂盒jetprime transfection reagent购自polyplus transfection公司。
94.2、sgrna设计与选择
95.在ncbi上检索出仓鼠polq基因及lig4基因,用snapgene选取该基因不同蛋白亚型上游的共有序列粘贴至crispr在线设计网站http://crispor.tefor.net/并生成多条sgrna,依据对两个基因sgrna特异性和有效性评分从高到低分别选择三组sgrna,并依据px330载体上的bbsi酶切位点,在选定的sgrna5’端加上caccg,对应互补链的3’端加上c,5’端加上aaac,设计出靶向polq基因的sgrna,其中,sgrna
‑
1、sgrna
‑
2、sgrna
‑
3的序列分别如
下所示:
96.sgrna
‑
1:5
’‑
atgagtcttccgcgccggagtgg
‑3’
(seq id no:1)
97.sgrna
‑
2:5
’‑
tccgactcgttctcgggagacgg
‑3’
(seq id no:2)
98.sgrna
‑
3:5
’‑
aaacggcggcgctcggcgtccgg
‑3’
(seq id no:3)
99.靶向rosa26位点的sgrna的序列如下所示:
[0100]5’‑
tcaagcgtgagcataaaactcgg
‑3’
(seq id no:4)
[0101]
px330质粒图谱见http://www.addgene.org/48138/。
[0102]
3、重组表达载体的构建与检测
[0103]
(1)sgrna
‑
px330载体的构建
[0104]
用bbsi限制性核酸内切酶在37℃、4h条件下对px330的bbsi位点进行特异性切割,配制1%琼脂糖凝胶并取5μl切割后的质粒电泳鉴定为特异线性质粒后直接进行柱纯化回收;同时将成对的sgrna稀释为10μm并各取22.5μl与10xtaq酶buffer混合成50μl的体系,在100℃下退火5min后自然冷却至室温形成双链,取退火后引物1μl、酶切后的px330质粒1μl、t4连接酶及t4连接酶buffer各1μl并用6μl的ddh2o补齐10μl,室温下进行连接(10min),42℃热激45s转化至trelief 5α感受态细胞,取100μl菌液涂布到氨苄抗性的lb琼脂糖固体培养基,37℃过夜培养,次日每块平板分别挑取5个菌落克隆,每个克隆分别加至5ml氨苄抗性的液体lb培养基中,37℃、200rpm过夜培养,次日每管取1ml送生工生物工程有限公司测序鉴定,余下保种,对于测序正确的重组质粒进行抽提、过滤;
[0105]
(2)t7ei验证载体基因编辑效率
[0106]
提前一天按2
×
105个细胞/孔将cho
‑
k1细胞接种于6孔板中,次日将6对sgrna
‑
px330载体与pmax
‑
gfp按4:1(4μg:1μg)共转至每孔细胞中,待转染48h后细胞汇合度为80%左右;流式分选富集gfp阳性细胞后用细胞基因组提取试剂盒提取细胞基因组,扩增polq基因,pcr的上游引物和下游引物的序列分别如下所示:
[0107]
上游引物:5
’‑
gcctctgggactgtgtcgtgt
‑3’
(seq id no:5)
[0108]
下游引物:5
’‑
cctgcgaccctcgatg
‑3’
(seq id no:6)
[0109]
pcr扩增产物柱纯化回收后进行梯度退火,用t7ei酶对退火产物进行酶切后,琼脂糖凝胶电泳检测切割条带,并根据切割带的灰度值验证载体编辑效率。
[0110]
4、实验结果
[0111]
cho细胞中针对polq的靶点序列及crispr/cas9载体图谱见图1,设计得到的靶向polq基因的sgrna的序列见图2,t7ei及ta克隆验证针对polq基因的crispr/cas9载体效率的结果图见图3,结果显示,靶向polq基因的sgrna2的编辑效率最高(38.4%),显著优于sgrna1和sgrna3,进一步将用sgrna2进行敲除的混合细胞克隆池基因组进行ta克隆测序,统计其中发生基因编辑效率,结果与t7ei验证的结果相符(39.6%)。
[0112]
实施例2构建polq基因敲除的cho
‑
k1细胞
[0113]
1、polq单基因敲除混合细胞池构建及单克隆细胞筛选
[0114]
提前一日按2
×
105个细胞将野生型cho
‑
k1铺至六孔板,选上述构建验证完成的高效sgrna
‑
px330(4μg)载体(sgrna2)与pmax
‑
gfp(1μg)质粒共转染cho
‑
k1细胞,48h后将细胞消化并用facs液清洗后过细胞筛网,流式细胞仪分选至含2%双抗及20%血清培养基的96孔板培养,每孔一个细胞,共制备20块具有单孔单个细胞的96孔板。至分选后第10天显微镜
下标记出具有单克隆细胞集落的孔并用胰酶消化转移至6孔板进行扩大培养,待孔中细胞汇合度达80%以上时,将细胞消化下来,2/3细胞用于冻存,1/3细胞继续培养2天后至汇合度为80%时提取基因组。
[0115]
2、polq单基因敲除单克隆细胞测序鉴定
[0116]
经一轮筛选获得共215株单克隆细胞,分批次对该组细胞进行鉴定:pcr扩增单克隆细胞基因组,pcr上游引物的序列如seq id no:5所示、下游引物的序列如seq id no:6所示。采用q5高保真dna聚合酶,参照说明书推荐的扩增程序用pcr仪进行扩增。扩增完毕后将产物以1%琼脂糖凝胶电泳进行纯化和胶回收目的片段;将胶回收纯化的pcr产物送生工生物股份有限公司直接测序,依据pcr产物测序峰图进行判断:当测序峰图出现明显的一大一小双峰结构则表示上机模板不止一个,即推测该产物包含至少一个等位基因突变,将所有具有双峰结构的单克隆pcr产物进一步进行ta克隆测序,将q5酶扩增的pcr产物经taq酶加a尾后通过对每个单克隆产物连接pmd
‑
19t载体,之后转化商业化感受态(top10)并挑取不少于30个ta克隆进行测序鉴定。对于每个单克隆细胞的30个ta克隆测序结果,应符合:(1)有且只有两种突变类型;(2)每种突变(无论插入或者删除)必须为移码突变,且蛋白翻译提前出现终止。经筛选鉴定得到三株单敲纯合细胞株。
[0117]
3、polq与lig4双敲细胞株构建
[0118]
在上述构建好的polq敲除细胞株基础上针对lig4进行敲除并进行鉴定,靶向lig4进行敲除的sgrna
‑
px330载体选择上述实施例1中t7ei验证后效率最高的载体,所述针对lig4基因的靶点序列如下所示:
[0119]5’‑
gcatgcagatgcacaaagatggg
‑3’
(seq id no:7)
[0120]
lig4敲除混合细胞池的构建参照本实施案例步骤1;单克隆细胞的获取及测序鉴定参照本实施步骤2,经一轮110株单克隆细胞筛选鉴定成功筛选出三株双敲纯合细胞株。
[0121]
4、western检测polq及lig4蛋白水平表达
[0122]
取对数生长期的野生型cho
‑
k1细胞及所获突变株接种于6孔板进行培养,待细胞生长至汇合度80%左右,用ripa裂解液提取细胞总蛋白,并用sds
‑
page蛋白电泳分离目的蛋白,电泳结束后将蛋白转印至pvdf膜上并用1
×
tbst进行清洗和封闭,之后用5%脱脂奶粉按1:500配制针对polq及lig4的一抗,4℃过夜封闭后次日用5%脱脂奶粉按1:2000配制辣根过氧化物酶标记的二抗并室温孵育1h,孵育完毕后1
×
tbst洗膜,用ecl显影。
[0123]
5、实验结果
[0124]
对polq基因敲除的cho
‑
k1单克隆细胞进行鉴定的结果图见图5a和图5b,对lig4基因单独敲除的结果图见图5c,polq与lig4双敲的cho单克隆细胞进行鉴定的结果图见图5d,结果显示,所述polq基因敲除的cho单克隆细胞为纯合的工具细胞株,所述lig4基因敲除的cho
‑
k1单克隆细胞为纯合的细胞株,所述polq与lig4双敲的cho
‑
k1单克隆细胞为纯合的细胞株。
[0125]
实施例3针对rosa26位点的基因敲入效率验证载体构建
[0126]
1、实验方法
[0127]
用noti、xhoi及sali各3μl对同源打靶骨架载体ploxp2neo(购自addgene公司)进行三酶切后进行去磷酸化(酶切4μg),1%琼脂糖凝胶电泳鉴定酶切条带位置并胶回收2.8kb条带;活化保存有rosa26
‑
sa
‑
egfp
‑
polya的大肠杆菌进行质粒小提,用noti及xhoi各
3μl对质粒进行双酶切并用1%琼脂糖凝胶回收1.9kb片段,t4 dna连接酶对目的片段及线性化载体进行连接后42℃热激45s转化至trelief 5α感受态细胞,取100μl菌液涂布到含氨苄抗性的lb琼脂糖固体培养基,37℃过夜培养,次日每块平板分别挑取5个菌落克隆,每个克隆分别加至5ml液体lb培养基中,37℃、200rpm过夜培养,次日每管取1ml送测序鉴定,余下保种,提取测序正确的重组质粒。
[0128]
2、实验结果
[0129]
结果显示,cho细胞中针对rosa26基因的靶点序列及同源重组效率验证载体图谱见图4,设计了缺少启动子并只有在整合到转录活性位点下游才能表达的带egfp的荧光报告载体,且该载体带有rosa26位点exon1区同源臂,仅当该基因整合到靶位点才能启动表达。
[0130]
实施例4工具细胞cho
‑
k1同源重组基因敲入效率验证
[0131]
1、实验方法
[0132]
取对数生长期的野生型cho
‑
k1细胞及所获突变株按2
×
105细胞每孔接种于6孔板进行培养,次日将靶向rosa26位点的sgrna
‑
px330(2μg)及rosa26
‑
sa
‑
egfp
‑
polya(6μg)同源重组效率验证载体共转染至上述细胞中,发生外源基因同源重组的细胞即表达gfp,72h后facs分析gfp阳性率并作比较;
[0133]
所述lig4基因单独敲除,以及polq与lig4双敲的cho
‑
k1单克隆细胞同源重组基因敲除效率的验证过程同上。
[0134]
2、实验结果
[0135]
流式分析同源重组效率的结果见图6a和图6b,结果显示,相比于野生型,经polq基因敲除的cho
‑
k1细胞的同源重组效率提升了35
‑
45倍,与polq
‑
/
‑
lig4
‑
/
‑
双敲的cho
‑
k1细胞的同源重组效率相当。而单独敲除lig4基因,同源重组效率仅提高了10倍左右。表明了单独敲除polq基因就能发挥主要的提高同源重组效率的效果,而敲除lig4基因的作用效果则较弱。
[0136]
实施例5工具细胞cho
‑
k1生长特性评价
[0137]
1、实验方法
[0138]
取对数生长期的野生型cho
‑
k1细胞及本发明构建得到的敲除polq基因的cho
‑
k1细胞突变株按2
×
105细胞每孔接种于6孔板进行培养,每过12h进行一次计数,并观察细胞生长形态。
[0139]
2、实验结果
[0140]
野生型cho
‑
k1细胞及本发明构建得到的敲除polq基因的cho
‑
k1细胞突变株的细胞生长曲线图见图7,结果显示,敲除polq基因的cho
‑
k1细胞的增殖能力相较于野生型而言无明显区别,表明了本发明构建得到的敲除polq基因的cho
‑
k1细胞的增殖能力不受影响。
[0141]
上述实施例的说明只是用于理解本发明的方法及其核心思想。应当指出,对于本领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也将落入本发明权利要求的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1