具有活性端的萘酰亚胺类化合物及其制备方法和应用
1.本发明属于精细化工技术领域,具体涉及一种具有活性端的萘酰亚胺类化合物及其制备方法和应用。萘酰亚胺类染料在荧光成像、多光子吸收、有机电子领域均具有重要的应用价值。本发明提出的聚集诱导发光性质的萘酰亚胺类化合物,提高了萘酰亚胺类染料在聚集状态下的荧光发光性质,使其在上述应用领域的性能得到进一步提高。
背景技术:
2.发光分子在聚集状态下普遍呈现荧光猝灭现象(aggregation
‑
caused quenching,acq),这极大地困扰了实际应用中发光分子在聚集态(薄膜、纳米离子)下的应用前景。香港科技大学唐本忠院士课题组2001年发现的聚集诱导发光(aggregation
‑
induced emission,aie)材料极大地改善了有机光电材料在上述器件中的性能,成为近年来有机光电材料发展的重要方向之一。
3.萘酰亚胺类荧光材料是著名的高荧光量子效率染料分子,在荧光探针、光致变色、双光子、电致发光、超分子自组装材料等领域得到长足发展。而该类分子由于平面刚性和强的分子内电荷转移能力,在聚集状态下容易出现π
‑
π堆积和扭曲的分子内电荷转移(twisted
‑
intramolecular charge transfer,tict),从而造成强的聚集诱导荧光猝灭现象。因此,应用于薄膜和纳米材料中往往需要通过物理掺杂和复杂的化学结构修饰来抑制聚集,而上述设计方案极大地增加了成本和环境污染,不符合现在“碳中和”概念。
4.通过荧光分子结构的微调,抑制分子聚集时的π
‑
π堆积、分子内和分子间的电子或电荷转移,从而有效提高分子辐射跃迁几率是将现有acq型荧光分子改造成aie型荧光分子的研究方向之一。其中,通过引入旋转、位阻单元,调节聚集态时的分子构象和堆积结构是该技术的一个重要技术手段。对于萘酰亚胺类荧光分子进行结构分析可以发现,其自身拥有一个刚性的稠环结构,缺少旋转单元。因此,引入合适结构和数量的旋转单元可以有效地改善其聚集状态。有研究表明,在萘酰亚胺的4
‑
位引入三键、双键、大位阻的酚和芳胺均可以构建具有aie性质的萘酰亚胺分子(mukherjee s,et.al.chemistry
‑
a european journal 2014,20(26):8012
‑
8023.gopikrishna p,et.al.acs applied materials&interfaces 2018,10(15):12081
‑
12111.)。而对其酰亚胺单元进行的aie改造与尝试并未得到重视(balachandra c.,et.al.j.org.chem.2020,85,3,1525
‑
1536)。由于4
‑
位同时具有改变发光波长的重要用途,将其应用于aie活性改造往往不利于其波长的进一步调控(delente,jm,et.al.chemical communication,2020,56(17):2562
‑
2565.qi q,et.al.chemical communication,2019,55(10):1446
‑
1449.)。因此,开发更加有效的aie调控策略和新型aie活性萘酰亚胺荧光团有着重要的技术开发意义。
5.应用于荧光成像的荧光分子,其靶向性修饰是一个重要的研究方向。而现有的aie活性萘酰亚胺荧光分子,大都不具备进一步靶向修饰的能力(zheng x,et al.chemical science 2019,10(8):2342
‑
2348.luo x,et.al.chemical engineering journal,2021,415:129095.)。因此,在上述技术开发的基础上,引入有效的活性单元,构建具有进一步靶
向修饰能力的荧光成像分子平台是对应用于荧光成像的萘酰亚胺分子的进一步技术要求。
技术实现要素:
6.针对现有技术中的不足,本发明的目的是提供一种具有活性端的聚集诱导发光萘酰亚胺类化合物及其制备方法和应用。其具有合成简单、反应条件温和、聚集态发光性能较好等特点,可以应用于细胞水平的荧光成像,并具备进一步靶向修饰的活性基。
7.为达到上述目的,本发明的解决方案是:
8.作为目的之一,本发明提供了一种具有活性端的萘酰亚胺类化合物,其具有式(ⅰ)所示的结构式:
[0009][0010]
其中,r选自酚或氨基醇。
[0011]
优选地,r选自羟基位于邻位、间位或对位的氨基苯酚、或c1
‑
c4氨基醇。
[0012]
作为目的之二,本发明提供了一种上述的具有活性端的萘酰亚胺类化合物的制备方法,其包括以下步骤:
[0013]
(a)、取nh2‑
(ch2)
m
‑
oh(m=1
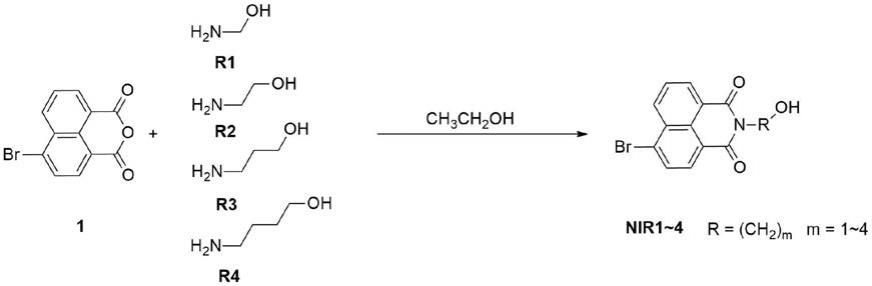
‑
4)或邻
‑
、间
‑
、对
‑
氨基苯酚和4
‑
溴
‑
1,8
‑
萘二甲酸酐溶解在乙醇中并进行加热反应,经第一次后处理得到酰胺上氮(n)取代的4
‑
溴
‑
1,8
‑
萘酰亚胺;
[0014]
(b)、取步骤(a)得到的酰胺上氮(n)取代的4
‑
溴
‑
1,8
‑
萘酰亚胺与9,9
‑
二甲基芴
‑2‑
硼酸溶解在溶剂中,在催化剂和碱液下进行加热反应,经第二次后处理得到最终的目标化合物。
[0015]
优选地,步骤(a)中,4
‑
溴
‑
1,8
‑
萘二甲酸酐、nh2‑
(ch2)
m
‑
oh和乙醇的摩尔体积比为1.0mmol:(1.1
‑
1.5)mmol:(15
‑
30)ml。
[0016]
优选地,步骤(a)中,4
‑
溴
‑
1,8
‑
萘二甲酸酐、邻
‑
、间
‑
、对
‑
氨基苯酚和乙醇的摩尔体积比为1.0mmol:(1.1
‑
1.5)mmol:(15
‑
30)ml。
[0017]
优选地,步骤(a)中,加热反应的温度为65
‑
85℃,加热反应的时间为6
‑
10h;第一次后处理具体为:待反应结束之后,冷却至室温,再加入水析出粗产物,抽滤得到的滤饼经真空干燥后用无水乙醇重结晶。
[0018]
优选地,步骤(b)中,酰胺上氮(n)取代的4
‑
溴
‑
1,8
‑
萘酰亚胺与9,9
‑
二甲基芴
‑2‑
硼酸、催化剂和溶剂的摩尔体积比为1.0mmol:(1.0
‑
1.1)mmol:(0.01
‑
0.02)mmol:(5
‑
10)ml,碱液的浓度为(1.3
‑
1.5)mol/l。
[0019]
优选地,步骤(b)中,溶剂选自四氢呋喃(thf)或n,n
‑
二甲基甲酰胺(dmf)。
[0020]
优选地,步骤(b)中,催化剂为四(三苯基膦)钯(pd(pph3)4)。
[0021]
优选地,步骤(b)中,碱液为碳酸钾(k2co3)溶液。
[0022]
优选地,步骤(b)中,加热反应的温度为60
‑
80℃,加热反应的时间为6
‑
12h;第二次
后处理具体为:待反应结束之后,冷却至室温,加入水析出粗产物,抽滤得到的滤饼溶解在二氯甲烷后用水萃取旋干,用无水乙醇重结晶。对纯度有较高要求的,可在重结晶后以100
‑
200目硅胶层析柱再次提纯。
[0023]
作为目的之三,本发明提供了一种上述的具有活性端的萘酰亚胺类化合物可以作为细胞荧光成像用造影剂的应用。可对细胞质进行成像,其活性端可引入亚细胞定位基团和探针识别单元。
[0024]
由于采用上述方案,本发明的有益效果是:
[0025]
第一、本发明的萘酰亚胺类化合物以1,8
‑
萘二甲酰胺为吸电子体,以9,9
‑
二甲基芴为给电子体,通过改变分子酰胺位的基团使之具有聚集诱导发光性能,该系列化合物在溶液中发光很弱,但在聚集状态下显示出强烈的绿色荧光,表现出明显的聚集诱导发光性质。同时,该系列化合物在不同极性的溶剂中显示出不同颜色的荧光,具有明显的溶剂效应。
[0026]
第二、本发明的制备方法具有合成简单、反应条件温和、聚集态发光性能较好等特点,可以应用于细胞水平的荧光成像,并具备进一步靶向修饰的活性基。
[0027]
第三、本发明提出的化合物结构具有羟基活性端,可以通过成酯、成醚反应引入探针识别单元、亚细胞定位单元等,实现在荧光探针、亚细胞成像等应用领域的拓展,故本发明的化合物可用于细胞成像。
附图说明
[0028]
图1为本发明的化合物nirf2在甲苯、四氢呋喃、二氯甲烷、乙腈、二甲基亚砜和乙醇中的紫外
‑
可见吸收光谱图。
[0029]
图2为本发明的化合物nirf2在甲苯、四氢呋喃、二氯甲烷、乙腈、二甲基亚砜和乙醇中的荧光发射光谱图(激发波长:375nm,激发/发射狭缝2/5nm)。
[0030]
图3为本发明的化合物nirf6在甲苯、四氢呋喃、二氯甲烷、乙腈、二甲基亚砜和乙醇中的紫外
‑
可见吸收光谱图。
[0031]
图4为本发明的化合物nirf6在甲苯、四氢呋喃、二氯甲烷、乙腈、二甲基亚砜和乙醇中的荧光发射光谱图(激发波长:370nm,激发/发射狭缝2/5nm)。
[0032]
图5为本发明的化合物nirf2在水含量为0%、10%、20%、30%、40%、50%、60%、70%、80%、90%、99%的四氢呋喃溶液中的荧光发射强度图(激发波长:375nm,激发/发射狭缝:2/5nm)。
[0033]
图6为本发明的化合物nirf2的不同水含量的荧光发射强度比例折线图。
[0034]
图7为本发明的化合物nirf6在水含量为0%、10%、20%、30%、40%、50%、60%、70%、80%、90%、99%的thf溶液中的荧光发射强度图(激发波长:370nm,激发/发射狭缝:2/5nm)。
[0035]
图8为本发明的化合物nirf6的不同水含量的荧光发射强度比例折线图。
[0036]
图9为本发明的化合物nirf6在水含量为50%的thf溶液中的动态光散射粒径
‑
荧光强度图。
[0037]
图10为本发明的化合物nirf6在水含量为99%的thf溶液中的动态光散射粒径
‑
荧光强度图。
[0038]
图11为本发明的化合物nir2的1h nmr谱图。
[0039]
图12为本发明的化合物nir6的1h nmr谱图。
[0040]
图13为本发明的化合物nirf2的1h nmr谱图。
[0041]
图14为本发明的化合物nirf2的
13
c nmr谱图。
[0042]
图15为本发明的化合物nirf6的1h nmr谱图。
[0043]
图16为本发明的化合物nirf6的
13
c nmr谱图。
[0044]
图17为本发明的化合物nirf2(10μmol/l)染色30min荧光成像图。
[0045]
图18为本发明的化合物nirf6(10μmol/l)染色30min荧光成像图。
具体实施方式
[0046]
本发明提供了一种具有活性端的萘酰亚胺类化合物及其制备方法和应用。
[0047]
<具有活性端的萘酰亚胺类化合物>
[0048]
本发明的具有活性端的萘酰亚胺类化合物的结构式如(ⅰ)所示:
[0049][0050][0051]
其中,r选自酚或氨基醇。酚具体为羟基位于邻位、间位或对位的氨基苯酚,氨基醇具体为c1
‑
c4氨基醇。
[0052]
<具有活性端的萘酰亚胺类化合物的制备方法>
[0053]
本发明的具有活性端的萘酰亚胺类化合物的制备方法包括以下步骤:
[0054]
(a)、取nh2‑
(ch2)
m
‑
oh(m=1
‑
4)或邻
‑
、间
‑
、对
‑
氨基苯酚和4
‑
溴
‑
1,8
‑
萘二甲酸酐溶解在乙醇中并进行加热反应,经第一次后处理得到酰胺上氮(n)取代的4
‑
溴
‑
1,8
‑
萘酰亚胺;
[0055]
(b)、取步骤(a)得到的酰胺上氮(n)取代的4
‑
溴
‑
1,8
‑
萘酰亚胺与9,9
‑
二甲基芴
‑2‑
硼酸溶解在溶剂中,在催化剂和碱液下进行加热反应,经第二次后处理得到最终的目标化合物。
[0056]
其中,在步骤(a)中,
[0057]
当r为氨基醇nh2‑
(ch2)
m
‑
oh时,具体的合成路线如下:
[0058]
[0059]
具体地,4
‑
溴
‑
1,8
‑
萘二甲酸酐、nh2‑
(ch2)
m
‑
oh(m=1
‑
4)和乙醇的摩尔体积比为1.0mmol:(1.1
‑
1.5)mmol:(15
‑
30)ml。
[0060]
当r为酚时,具体的合成路线如下:
[0061][0062]
具体地,4
‑
溴
‑
1,8
‑
萘二甲酸酐、邻
‑
、间
‑
、对
‑
氨基苯酚和乙醇的摩尔体积比为1.0mmol:(1.1
‑
1.5)mmol:(15
‑
30)ml。
[0063]
在步骤(a)中,加热反应的温度为65
‑
85℃,加热反应的时间为6
‑
10h,第一次后处理具体为:待反应结束之后,冷却至室温,再加入水析出粗产物,抽滤得到的滤饼经真空干燥后用无水乙醇重结晶。
[0064]
其中,在步骤(b)中,
[0065]
得到的中间体nir1
‑
nir7分别与9,9
‑
二甲基芴
‑2‑
硼酸进行suzuki反应,具体的合成路线如下:
[0066]
[0067]
具体地,在步骤(b)中,酰胺上氮(n)取代的4
‑
溴
‑
1,8
‑
萘酰亚胺(nir1
‑
nir7)与9,9
‑
二甲基芴
‑2‑
硼酸、催化剂和溶剂的摩尔体积比为1.0mmol:(1.0
‑
1.1)mmol:(0.01
‑
0.02)mmol:(5
‑
10)ml,碱液的浓度为(1.3
‑
1.5)mol/l。
[0068]
在步骤(b)中,溶剂选自thf或dmf,催化剂为pd(pph3)4,碱液为k2co3溶液。
[0069]
在步骤(b)中,加热反应的温度为60
‑
80℃,加热反应的时间为6
‑
12h;第二次后处理具体为:待反应结束之后,冷却至室温,加入水析出粗产物,抽滤得到的滤饼溶解在二氯甲烷后用水萃取旋干,用无水乙醇重结晶。对纯度有较高要求的,可在重结晶后以100
‑
200目硅胶层析柱再次提纯。
[0070]
其中,化合物nirf2与化合物nirf6在甲苯、四氢呋喃、二氯甲烷、乙腈、二甲基亚砜和乙醇中的最大吸收(λ
abs
.)、最大发射(λ
em
.)、摩尔消光系数(ε*λ
abs
.)、斯托克斯位移(
△
λ)如下表1所示:
[0071]
表1 化合物nirf2与化合物nirf6在不同溶剂中的光学性质
[0072][0073][0074]
由表1可知,不论r取代基为脂肪基还是芳香基,式(1)的化合物在水中均具有较好的荧光发光效率和较大的斯托克斯(stokes)位移。
[0075]
化合物nirf2与化合物nirf6的细胞成像照片说明,不论r取代基为脂肪基还是芳香基,式(1)的化合物均可以用于细胞成像。成像的激发波长可为405nm和458nm,优选405nm,收集的区间覆盖450
‑
750nm,优选500
‑
600nm。
[0076]
由图1可知,化合物nirf2在不同极性溶剂中,紫外吸收光谱在369
‑
375nm的狭窄范围内,说明通过氟原子的引入,使得萘酰亚胺分子内出现弱化的分子内电荷转移,从而吸收光谱显示出极小的波长移动。
[0077]
由图2可知,化合物nirf2在不同极性溶剂中,荧光发射光谱在464
‑
529nm范围内。随着溶剂极性的增加,其荧光出现多达65nm的红移,说明萘酰亚胺分子激发态的电荷转移性质受到溶剂极性影响较大。
[0078]
由图3可知,化合物nirf6在不同极性溶剂中,紫外吸收光谱在365
‑
372nm的狭窄范围内,说明通过氟原子的引入,使得萘酰亚胺分子内出现弱化的分子内电荷转移,从而吸收光谱显示出极小的波长移动。与化合物nirf2相比,化合物nirf6引入的刚性苯环使得材料吸收光谱出现极微小的蓝移,这是由于苯环并未参与分子发色团的共轭,对其前线轨道影响较小。
[0079]
由图4可知,化合物nirf6在不同极性溶剂中,荧光发射光谱在470
‑
526nm范围内。随着溶剂极性的增加,其荧光出现多达56nm的红移,说明萘酰亚胺分子激发态的电荷转移性质受到溶剂极性影响较大。同样,与化合物nirf2的发射光谱差异不大,这也是由于在有机溶剂中,荧光主要来自非聚集态的分子发光所致。
[0080]
由图5可知,含水量在0
‑
10%区间荧光增强,在10
‑
80%时,荧光强度逐渐降低,在80
‑
99%区间,荧光强度逐渐升高;说明化合物nirf2在含水量低的时候发生聚集诱导猝灭,当聚集达到一定程度后,产生聚集体发光,发光性能有所增强。
[0081]
图6详细描绘了图5中荧光随体系含水量变化而产生的聚集诱导增强效果。图6说明具有柔性链的化合物rinf2的聚集诱导发光现象并不明显,在较高含水量区间的荧光增强倍数也较低。
[0082]
由图7可知,含水量在0
‑
50%时,荧光强度逐渐增强,并在50
‑
60%区间保持最强的荧光发射,当含水量超过60%时,由于化合物nirf6聚集尺寸增加,分散性降低,荧光逐渐减弱;表明了化合物nirf6的聚集诱导发光性质。
[0083]
图8详细描绘了图7中荧光随体系含水量变化而产生的聚集诱导增强效果。图8说明具有额外刚性苯环的化合物rinf6的聚集诱导发光现象较化合物nirf2大幅提高,随着含水量增加,荧光强度呈直线上升趋势,直至含水量在50
‑
60%处达到峰值。
[0084]
由图9可知,化合物nirf6的水合粒径在含水量50%处达到141.772nm,证明溶液中形成了亚稳态纳米聚集体。
[0085]
由图10可知,随着含水量的增加,当含水量增加到99%时,化合物nirf6的水合粒径达到341.995nm,较大的尺寸造成了化合物在该状态下较差的分散性。
[0086]
由图17可知,化合物nirf2在细胞中均匀分布,且具有一定的荧光强度,共聚焦荧光显微镜照片较清晰。
[0087]
由图18可知,化合物nirf6在细胞中均匀分布,且荧光强度明显较化合物nirf2更高,共聚焦荧光显微镜照片更加清晰。说明引入刚性非共轭的苯环对这一类萘酰亚胺的聚集诱导发光性质有极大的提高,并且这一性能在细胞层面的成像过程中得以保持,极大地提高了成像的灵敏度。
[0088]
<具有活性端的萘酰亚胺类化合物的应用>
[0089]
本发明的具有活性端的萘酰亚胺类化合物可以作为细胞荧光成像用造影剂的应用。可对细胞质进行成像,其活性端可引入亚细胞定位基团和探针识别单元。
[0090]
下面结合实施例对本发明的技术内容做进一步的说明。下述实施例是说明性的,不是限定性的,不能以下述实施例来限定本发明的保护范围。下述实施例中所使用的实验
方法如无特殊说明,均为常规方法。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0091]
以下结合实施例对本发明作进一步的说明。
[0092]
实施例1:化合物nir2的合成
[0093][0094]
在一个配有磁性搅拌子的250ml三口烧瓶中,依次加入4
‑
溴
‑
1,8
‑
萘二甲酸酐(化合物1)(2.77g,10mmol)、乙醇胺(670mg,11mmol)以及150ml乙醇,升温至85℃,搅拌回流6h。自然冷却至室温,倾倒入含200ml水的烧杯中搅拌,之后静置并抽滤得到滤饼,用饱和nahco3溶液洗涤后真空干燥,用200ml无水乙醇重结晶,得到2.75g浅褐色粉末nir2,产率为86%。1h nmr(400mhz,dmso
‑
d6)δppm 3.58
‑
3.66(m,2h),4.14(t,j=6.53hz,2h),4.86(t,j=6.02hz,1h),8.00(t,j=7.91hz,1h),8.22(d,j=8.03hz,1h),8.32(d,j=7.78hz,1h),8.52
‑
8.59(m,2h)。
[0095]
实施例2:化合物nirf2的合成
[0096][0097]
室温条件下,将nir2(1.6g,5mmol)、9,9
‑
二甲基芴
‑2‑
硼酸(1.19g,5mmol)、
[0098]
pd(pph3)4(57mg,0.05mmol)以及25ml的dmf置于50ml反应管中。然后加入k2co3水溶液5ml(k2co3溶液的浓度为1.30mol/l),使固体化合物完全溶解,得到棕红色透明溶液,鼓泡脱除氧气,氮气保护,逐渐升温至80℃,反应6h。自然冷却至室温,倾倒入100ml水中,抽滤得到的滤饼溶解二氯甲烷(20ml),饱和食盐水(20ml
×
2)洗涤后收集合并有机相,无水na2so4干燥,旋干后粗产物用200ml乙醇重结晶,得1.95g的白色产物nirf2,收率为90%。如需进一步提纯,则用100
‑
200目硅胶层析柱提纯(洗脱剂为二氯甲烷),得到1.77g的白色产物nirf2,产率为82%。1hnmr(400mhz,cdcl3)ppm 1.59(s,6h),2.51(t,j=5.52hz,1h),4.05(m,2h),4.54(t,j=5.14hz,2h),7.40
‑
7.45(m,2h),7.49
‑
7.54(m,2h),7.60(d,j=0.75hz,1h),7.73
‑
7.78(m,1h),7.81
‑
7.86(m,2h),7.91(d,j=7.78hz,1h),8.42(d,j=8.28hz,1h),8.70(t,j=1.00hz,2h)。
13
c{h}nmr(100mhz,cdcl3)ppm 27.17,42.90,47.12,53.42,61.97,120.10,120.37,121.24,122.60,122.76,124.23,126.88,127.24,127.90,128.00,128.89,129.05,130.22,131.21,131.53,133.20,137.54,138.38,139.77,147.87,153.93,154.22,165.10,165.29。
[0099]
实施实例3:化合物nir6的合成
[0100][0101]
在一个配有磁性搅拌子的250ml三口烧瓶中,依次加入4
‑
溴
‑
1,8
‑
萘二甲酸酐(化合物1)(2.77g,10mmol)、间氨基苯酚(1.09g,11mmol)以及150ml乙醇,升温至85℃,搅拌回流6h。自然冷却至室温,倾倒入含200ml水的烧杯中搅拌,之后静置并抽滤得到滤饼,用饱和nahco3溶液洗涤后真空干燥,干燥后用200ml无水乙醇重结晶,得到3.1g白色粉末nir6,产率为84%。1h nmr(400mhz,dmso
‑
d6)δppm:9.67(s,1h),8.58(s,1h),8.61(dd,2h),8.34
‑
8.36(d,j=7.48,1h),8.26(d,j=8.23,1h),8.02
‑
8.05(m,1h),7.29
‑
7.32(m,1h),6.85
‑
6.87(m,1h),6.79(m,2h)。
[0102]
实施实例4:化合物nirf6的合成
[0103][0104]
室温条件下,将nir6(1.84g,5mmol)、9,9
‑
二甲基芴
‑2‑
硼酸(1.19g,5mmol)、
[0105]
pd(pph3)4(57mg,0.05mmol)以及25ml的dmf置于50ml反应管中。然后加入k2co3水溶液5ml(k2co3溶液的浓度为1.30mol/l),使固体化合物完全溶解,得到棕红色透明溶液,鼓泡脱除氧气,氮气保护,逐渐升温至80℃,反应6h。自然冷却至室温,倾倒入100ml水中,抽滤得到的滤饼溶于二氯甲烷(20ml),饱和食盐水(20ml
×
2)洗涤后收集合并有机相,无水na2so4干燥,旋干后粗产物用200ml乙醇重结晶,得2.21g的白色产物nirf6,收率为91%。如需进一步提纯,则用100
‑
200目硅胶层析柱提纯(洗脱剂为二氯甲烷),得到2.03g的白色产物nirf6,产率为84%。1hnmr(400mhz,cdcl3)ppm 1.59(s,6h)2.51(t,j=5.52hz,1h),4.05(m,j=5.30,5.30,5.30hz,2h),4.54(t,j=5.14hz,2h),7.40
‑
7.45(m,2h),7.49
‑
7.54(m,2h),7.60(d,j=0.75hz,1h),7.73
‑
7.78(m,1h),7.81
‑
7.86(m,2h),7.91(d,j=7.78hz,1h),8.42(d,j=8.28hz,1h),8.70(t,j=1.00hz,2h)。
13
c{h}nmr(100mhz,cdcl3)ppm 47.12,116.13,116.29,117.29,120.12,120.37,120.59,121.56,122.76,122.93,124.20,126.93,127.25,127.91,128.06,129.02,129.21,130.34,131.40,131.73,133.30,136.27,137.56,138.38,139.83,147.36,148.01,153.93,154.26,156.95,164.36,164.56。
[0106]
实施实例5:化合物nirf2和化合物nirf6的细胞成像实验。
[0107]
(a)、配制化合物nirf2和化合物nirf6的母液(1mmol/l)。
[0108]
(b)、在14mm爬片上预先孵育hela细胞24h,移除培养液,用pbs缓冲液冲洗共聚焦培养皿3次,加入990ml新的培养基,取化合物(nirf2或nirf6)母液10ml加入共聚焦培养皿,继续在37℃,5%的co2条件下孵育30min。之后用pbs缓冲液冲洗共聚焦培养皿3次,加入2ml的pbs缓冲液。
[0109]
(c)、在共聚焦显微镜下,设置激发光源波长为405nm,激光功率为40%。收集通道为:500
‑
600nm。
[0110]
上述对实施例的描述是为了便于该技术领域的普通技术人员能理解和使用本发
明。熟悉本领域技术人员显然可以容易的对这些实施例做出各种修改,并把在此说明的一般原理应用到其他实施例中,而不必经过创造性的劳动。因此,本发明不限于上述实施例。本领域技术人员根据本发明的原理,不脱离本发明的范畴所做出的改进和修改都应该在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1