一种亚精胺的制备方法与流程
1.本发明涉及有机化学合成领域,特别涉及以低成本、温和反应条件下实现高纯亚精胺工业化生产纯化及工业化生产的方法,尤其是一种亚精胺的制备方法。
背景技术:
2.亚精胺是一种多胺,广泛分布在生物体内,可抑制神经元no合成酶,结合并沉淀dna,也可用于纯化dna结合蛋白,刺激t4聚核苷酸激酶活性。参与生物体内许多的生物学过程,如调控细胞增殖、细胞衰老、器官发育、免疫以及癌症等生理和病理过程。
3.2013年9月1日,德国和奥地利的科学家合作研究表示,亚精胺或可阻止老年痴呆症发病。目前亚精胺除了从小麦胚芽中提取以外,也可通过化学合成的方法来获得,但是现有技术中的化学合成的方法中存在诸多问题。
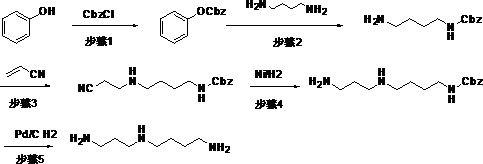
4.例如:天津市普莱克医药科技有限公司2012年申请专利申请号为cn201210138708.4,专利名称为:一种亚精胺的合成方法发明专利申请中提出一种以苄氧羰基为保护基的方法制备亚精胺,共有5步反应,首先制备苄氧羰基保护的1,4
‑
丁二胺,然后将其与丙烯腈进行加成反应,最后将加成产物中的氰基还原,并脱除苄氧羰基保护,得到亚精胺,总收率30%,产品纯度为99.5%。此方法后两步分别使用了雷尼镍和昂贵的钯碳来高压加氢还原,反应条件需要用到特种设备,生产成本高,其具体反应式如下:。
5.再如:四川大学在2018年申请的专利申请号为cn201810833825.x,专利名称为:制备亚精胺的方法的发明专利申请中也提供了一种制备亚精胺的方法,其包括以下步骤:a、以化合物ⅰ氨基丙醇和丁内酯为原料在溶剂中反应;b、化合物 经四氢锂铝还原得到化合物;c、化合物加入碱和二碳酸二叔丁酯反应以对羟基进行保护得到化合物;d、化合物加入配体和邻苯二甲酰亚胺反应得到化合物;e、化合物v溶于溶剂中,加入水合肼进行反应,反应结束,经后处理得到化合物vi,进一步精馏化合物vi,得到亚精胺纯品,总收率为35%,产品纯度为98%。经分析可以看出,该方法以氨基丙醇和丁内酯为原料反应后,经过还原、氨基保护等步骤得到亚精胺。此方法共五步,还原酰胺的步骤中使用到危险的试剂氢化铝锂,不易放大生产;另外,邻苯二甲酰亚胺引入氨基后需用水合肼脱除,反应安全风险高,氨基丙醇等原料均较为昂贵,整体成本较高,其具体反应式如下:
。
6.当然,现有的亚精胺制备方法并不仅限于上述两种,但是,现有的亚精胺制备方法通常是存在上述示例中的原料成本高、反应条件要求苛刻、存在安全隐患等问题的。
7.因此,目前亟需提出一种能够以低成本、便捷的制备路径实现亚精胺制备的新方法,用以更好地解决现有技术中存在的问题。
技术实现要素:
8.本发明为解决上述技术问题之一,本发明的目的在于:提出一种以易得的原料、较低的成本及温和的反应条件,解决高纯亚精胺工业化生产的纯化及易放大后实现工业化生产的问题的技术方案。
9.为实现本发明的发明目的,其所采取的具体技术方案是:一种亚精胺的制备方法,所述亚精胺的分子式为c7h
19
n3,化学结构式如下:,包括如下步骤:s1、以4
‑
二甲氨基吡啶(dmap)为催化剂,吡咯烷酮(化合物a)和二碳酸二叔丁酯在有机溶剂中反应,反应完全后脱溶,经后处理后得到化合物b,其具体反应式如下:;s2、将化合物b、1,3
‑
丙二胺在有机溶剂中搅拌反应至反应结束,经弱酸水溶液萃取、有机溶剂洗杂、碱调节ph、加盐析、有机溶剂萃取产物、减压脱溶得到化合物c,其具体反应式如下:;s3、化合物c溶于有机溶剂中,经还原剂还原,再经淬灭、洗涤等后处理步骤得到化合物d,其具体反应式如下:;
s4、化合物d溶于溶剂,经酸催化,反应脱去保护基,反应结束后经碱调节ph,进一步处理得到未经纯化的化合物e,进一步经减压精馏得到亚精胺纯品,其具体反应式如下:。
10.在上述任一方案中,作为优选的是,上述步骤s1中,所述的反应步骤使用的有机溶剂可以为乙腈、四氢呋喃、甲基叔丁基醚等常用的单一有机溶剂或者混合溶剂;进一步地,如果使用甲基叔丁基醚等水不相溶的溶剂可无需减压脱溶直接进入后处理水洗步骤。
11.在上述任一方案中,作为优选的是,上述步骤s1中,反应温度包括于10℃至20℃或者20℃至30℃,反应时间为2小时至10小时。
12.在上述任一方案中,作为优选的是,上述步骤s1中,所述的后处理方法为依次经弱酸水溶液洗涤、弱碱水溶液洗涤,减压脱溶至干,所述的弱酸包括柠檬酸、氯化铵;弱碱包括碳酸氢钠、碳酸钠、碳酸氢钾。
13.在上述任一方案中,作为优选的是,上述步骤s1中,所述的催化剂4
‑
二甲氨基吡啶(dmap)和吡咯烷酮的摩尔比例为1:5至1:20。
14.在上述任一方案中,作为优选的是,上述步骤s1中,所述的2
‑
吡咯烷酮和二碳酸二叔丁酯的摩尔比例为5:5至5:6。
15.在上述任一方案中,作为优选的是,上述步骤s2中,所述的溶剂为四氢呋喃、乙腈、正庚烷、正己烷、石油醚中的任意一种或者多种的任意混合溶剂。
16.在上述任一方案中,作为优选的是,上述步骤s2中,所述的化合物b 1
‑
(叔丁氧基羰基)
‑2‑
吡咯烷酮与1,2
‑
丙二胺的摩尔比为5:6至5:10。
17.在上述任一方案中,作为优选的是,上述步骤s2中,所述的反应步骤使用的有机溶剂包括乙腈、四氢呋喃、正庚烷、正己烷、石油醚,洗杂的有机溶剂包括二氯甲烷、乙酸乙酯、醋酸异丙酯等,萃取产物所用的有机溶剂包括为2
‑
甲基四氢呋喃、二氯甲烷、正丁醇。
18.在上述任一方案中,作为优选的是,上述步骤s2中,所述的弱酸水溶液包括柠檬酸水溶液、氯化铵水溶液。
19.在上述任一方案中,作为优选的是,上述步骤s3中,至少满足以下任意一项:所述的溶剂为四氢呋喃或者甲苯;所述的还原剂为硼烷四氢呋喃、硼烷二甲硫醚络合物、路易斯酸和硼氢化钠组合及红铝中的任意一种,其中,所述的路易斯酸包括三氟化硼、氯化锌、氯化铝、氯化镁中的任意一种。
20.在上述任一方案中,作为优选的是,上述步骤s3中,所述化合物c与还原剂摩尔比为10:11至10:25。
21.在上述任一方案中,作为优选的是,上述步骤s4中,至少满足以下任意一项:所述有机溶剂为甲醇或者乙醇或其混合形成的混合溶剂;所述碱调节ph步骤中调节ph的碱选用氢氧化钠、氢氧化钾、碳酸钾、强碱性阴离子交换树脂中的一种。
22.在上述任一方案中,作为优选的是,上述步骤s4中,所述的酸包括盐酸、三氟乙酸。
23.在上述任一方案中,作为优选的是,上述步骤s4中,所述的进一步处理的方法是经脱溶、过滤方法或者树脂吸附除盐,再经脱溶步骤。
24.在上述任一方案中,作为优选的是,上述步骤s4中,所述减压精馏的条件为蒸馏外温严格控制142℃,在控制12mmhg真空下收集124℃至126℃馏分。
25.与现有技术相比,本发明的有益效果如下:1、本发明所使用原料吡咯烷酮、1,3
‑
丙二胺等均为常见化工原料,价格便宜易得;化工原料成本较低、反应条件相对温和,整体反应中安全隐患较低。
26.2、s1中经叔丁氧羰基保护,反应收率理想,可达94.2%至98.8%。
27.3、s2中经过量的1,3
‑
丙二胺开环吡咯烷酮,得到亚精胺的主链结构化合物c,在弱酸条件下产物易溶于水,可经有机溶剂洗涤分液的方式除去小极性杂质,进一步调节ph后盐析方式降低化合物c在水中的溶解度,经有机溶剂萃取后减压蒸馏除去大部分的丙二胺,提纯方法简单有效,单步收率范围为62.2%至73.2%。
28.4、s3中在还原剂存在下还原酰胺得到单boc保护的亚精胺。
29.5、s4中经酸催化脱去叔丁氧羰基保护基团,精馏后得到亚精胺产物。
30.6、利用本方法制得产物的合并收率范围达到62.1%~79.5%,且成品亚精胺的纯度可达到99.6%。
具体实施方式
31.下面将对本发明技术方案的实施例进行详细的描述。以下实施例仅用于更加清楚地说明本发明的技术方案,因此只作为示例,而不能以此来限制本发明的保护范围。
32.利用本发明中的方法实现亚精胺制备时,亚精胺的合成路径如下:。
33.实施例1:s1、合成下式的化合物b:1
‑
(叔丁氧基羰基)
‑2‑
吡咯烷酮,其分子式为:c9h
15
no3, 分子量为:185.22,化学结构式如下:;其制备方法包括如下步骤:将吡咯烷酮(255.1g,3.0mol,1.0eq),乙腈2.0l,4
‑
二甲氨基吡啶(18.3g,0.15mol,0.05eq)加入3l反应瓶中,水浴中滴加二碳酸二叔丁酯(654.7g,3.0mol,1.0eq),20~30℃反应5h,40℃减压浓缩干,加入1175g 甲基叔丁基醚溶解,用137g柠檬酸/1400ml水溶液,40g碳酸氢钠/600ml水溶液,40g氯化钠/600ml水溶液洗后,有机相脱除溶剂,经后处理提纯后得到浅黄色油状物化合物b共529.5g,收率95.3%;s2、合成下式的化合物c:[3
‑
(3
‑
氨基
‑
丙氨甲酰基)丙基]
‑
氨基甲酸叔丁酯,其分
子式为:c
12
h
25
n3o3,分子量为:259.35,化学结构式如下:;其制备方法包括如下步骤:将1,3
‑
丙二胺(80.0g,1.08mol)、化合物b(100g,0.54mol)及乙腈400ml混合,于20~25℃搅拌24h,减压脱除溶剂和过量的1,3
‑
丙二胺,剩余体系加入400ml水中,用柠檬酸调至ph=5,二氯甲烷m 100ml萃取4次,水层加入60g氯化钠,用30%氢氧化钠水溶液调至ph=10,水相用2
‑
甲基四氢呋喃150ml*2萃取,脱除溶剂,得到黄色油状物(化合物c)102.5g,收率73.2%;s3、合成下式的化合物d:[4
‑
(3
‑
氨基
‑
丙氨基)丁基]
‑
氨基甲酸叔丁酯,其分子式为:c
12
h
27
n3o2,分子量为:245.36,化学结构式如下:;其制备方法包括如下步骤:将化合物c(78g,0.3mol)溶于四氢呋喃1000ml,氮气保护,冰浴下0
±
5℃滴加50%硼烷四氢呋喃溶液(168g,0.6mol),升至30℃,保温16h,冰浴下滴入甲醇600ml淬灭反应,45℃水浴减压浓缩干,得化合物d;s4、合成下式的化合物e:n
‑
(3
‑
氨基丙基)
‑
1,4
‑
丁二胺,即亚精胺,其分子式为:c7h
19
n3,分子量为:145.25,化学结构式如下:;其制备方法包括如下步骤:将上一步化合物d(0.3mol)溶于450ml甲醇中,冰水浴中滴入盐酸(250ml,3mol),搅拌4h,减压脱去甲醇,用40%氢氧化钠水溶液调至ph=12,减压脱干,加入四氢呋喃300ml萃取,过滤,滤液浓缩干,进一步减压精馏,于12mmhg真空下收集124~126℃馏分,得到化合物e,即亚精胺34.6g,收率79.5%,气相纯度99.5%。
[0034]
实施例2:s1、合成下式的化合物b:1
‑
(叔丁氧基羰基)
‑2‑
吡咯烷酮,其分子式为:c9h
15
no3,分子量为:185.22,化学结构式如下:;其制备方法包括如下步骤:将吡咯烷酮(255.1g,3.0mol,1.0eq),四氢呋喃1.8l,4
‑
二甲氨基吡啶(18.3g,0.15mol,0.05eq)加入3l反应瓶中,水浴中滴加二碳酸二叔丁酯(720.2g,3.3mol,1.1eq),20~30℃反应5h,40℃减压浓缩干,加入1175g甲基叔丁基醚溶解,用137g柠檬酸/1400ml水溶液,40g 碳酸氢钠/600ml水溶液,40g氯化钠/600ml水溶液洗后,有机相脱除溶剂,经后处理提纯后得到浅黄色油状物化合物b共533.9g,收率96.1%;s2、合成下式的化合物c:[3
‑
(3
‑
氨基
‑
丙氨甲酰基)丙基]
‑
氨基甲酸叔丁酯,其分子式为:c
12
h
25
n3o3,分子量为:259.35,化学结构式如下:;其制备方法包括如下步骤:将1,3
‑
丙二胺(60.0g,0.81mol,1.5eq)、化合物b(100g,0.54mol,1.0eq)及正庚烷700ml混合,于20~25℃搅拌30h,减压脱除溶剂和过量的1,3
‑
丙二胺,剩余体系加入400ml水中,用柠檬酸调至ph=5,乙酸乙酯100ml萃取3次,水层加入60g氯化钠,用30%氢氧化钠水溶液调至ph=10,水相用2
‑
甲基四氢呋喃150ml*2萃取,脱除溶剂,得到黄色油状物(化合物c)95.2g,收率68.0%;s3、合成下式的化合物d:[4
‑
(3
‑
氨基
‑
丙氨基)丁基]
‑
氨基甲酸叔丁酯,其分子式
为:c
12
h
27
n3o2,分子量为:245.36,化学结构式如下:;其制备方法包括如下步骤:将化合物c(78g,0.3mol)溶于四氢呋喃1000ml,氮气保护,冰浴下控温0
±
5℃滴加10n硼烷二甲硫醚络合物(60ml,0.6mol),自然升至30℃,保温16h,冰浴下滴入甲醇600ml淬灭反应,45℃水浴减压浓缩干,得化合物d;s4、合成下式的化合物e:n
‑
(3
‑
氨基丙基)
‑
1,4
‑
丁二胺,即亚精胺,其分子式为:c7h
19
n3,分子量为:145.25,化学结构式如下:;其制备方法包括如下步骤:将上一步化合物d(0.3mol)溶于450 ml甲醇中,冰水浴中滴入盐酸(250ml,3mol),搅拌4h,减压蒸干,加入纯化水300克,经d201强碱性阴离子树脂柱洗脱,60℃下减压浓缩至干,四氢呋喃洗脱产物,过滤,滤液减压脱溶后剩余物精馏,于12mmhg真空下收集124~126℃馏分,得到亚精胺32.8g,收率75.5%,气相纯度99.6%。
[0035]
实施例3:s1、合成下式的化合物b:1
‑
(叔丁氧基羰基)
‑2‑
吡咯烷酮,其分子式为:c9h
15
no3,分子量为:185.22,化学结构式如下:;其制备方法包括如下步骤:将吡咯烷酮(255.1g,3.0mol,1.0eq),甲基叔丁基醚2.0l,4
‑
二甲氨基吡啶(36.65g,0.3mol,0.1eq)加入3l反应瓶中,水浴中滴加二碳酸二叔丁酯(720.2g,3.3mol,1.1eq),20~30℃反应3h,加入250g氯化铵/1400ml水溶液,40g碳酸氢钠/600ml水溶液,有机相脱除溶剂,经后处理提纯后得到浅黄色油状物化合物b共523.4g,收率94.2%;s2、合成下式的化合物c:[3
‑
(3
‑
氨基
‑
丙氨甲酰基)丙基]
‑
氨基甲酸叔丁酯,其分子式为:c
12
h
25
n3o3,分子量为:259.35,化学结构式如下:;其制备方法包括如下步骤:将1,3
‑
丙二胺(80.0g,1.08mol,2.0eq)、化合物b(100g,0.54mol,1.0eq)及四氢呋喃400ml混合,于室温搅拌30h,减压脱除溶剂和过量的1,3
‑
丙二胺,剩余体系加入400ml水中,用柠檬酸调至ph=5,二氯甲烷100ml萃取4次,水层加入60g氯化钠,用30%氢氧化钠水溶液调至ph=10,水相用2
‑
甲基四氢呋喃150ml*2萃取,脱除溶剂,得到黄色油状物(化合物c)91.0g,收率65.0%;s3、合成下式的化合物d:[4
‑
(3
‑
氨基
‑
丙氨基)丁基]
‑
氨基甲酸叔丁酯,其分子式为:c
12
h
27
n3o2,分子量为:245.36,化学结构式如下:;其制备方法包括如下步骤:将化合物c(78g,0.3mol)溶于四氢呋喃1000ml,氮气保护,加入硼氢化钠(17.0g,0.45mol),冰浴下控温0
±
5℃滴加三氟化硼四氢呋喃(168g,0.6mol),加完后继续保温搅拌2小时,升温至40℃,保温12h,冰浴下滴入甲醇600ml淬灭反应,45℃水浴减压浓缩干,得化合物d;s4、合成下式的化合物e:n
‑
(3
‑
氨基丙基)
‑
1,4
‑
丁二胺,即亚精胺,其分子式为:c7h
19
n3,分子量为:145.25,化学结构式如下:;其制备方法包括
如下步骤:将上一步化合物d(0.3mol)溶于450ml甲醇中,冰水浴中滴入盐酸(250ml,3mol),搅拌4h,减压蒸干,用氢氧化钾调至ph=12,40℃搅拌2小时后,减压脱干,加入四氢呋喃300ml萃取,过滤,滤液浓缩干,减压精馏,于12mmhg真空下收集124~126℃馏分,得到亚精胺32.1g,收率73.8%,气相纯度99.1%。
[0036]
实施例4:s1、合成下式的化合物b:1
‑
(叔丁氧基羰基)
‑2‑
吡咯烷酮,其分子式为:c9h
15
no3,分子量为:185.22,化学结构式如下:;其制备方法包括如下步骤:将吡咯烷酮(255.1g,3.0mol,1.0eq),乙腈2.0l,4
‑
二甲氨基吡啶(73.3g,0.6mol,0.2eq)加入3l反应瓶中,水浴中滴加二碳酸二叔丁酯(654.7g,3.0mol,1.0eq),10~20℃反应2h,40℃减压浓缩干,加入1175g甲基叔丁基醚溶解,用137g柠檬酸/1400ml水溶液,40g碳酸氢钠/600ml水溶液,40g氯化钠/600ml水溶液洗后,有机相脱除溶剂,经后处理提纯后得到浅黄色油状物化合物b共540.1g,收率97.2%;s2、合成下式的化合物c:[3
‑
(3
‑
氨基
‑
丙氨甲酰基)丙基]
‑
氨基甲酸叔丁酯,其分子式为:c
12
h
25
n3o3,分子量为:259.35,化学结构式如下:;其制备方法包括如下步骤:将1,3
‑
丙二胺(80.0g,1.08mol,2.0eq),化合物b(100g,0.54mol,1.0eq)以及四氢呋喃500ml混合,于室温搅拌30h,减压脱除溶剂和过量的1,3
‑
丙二胺,剩余体系加入400ml水中,用柠檬酸调至ph=5,醋酸异丙酯100ml萃取3次,水层加入60g氯化钠,用30%氢氧化钠水溶液调至ph=7,水相用二氯甲烷150ml*4萃取,脱除溶剂,得到黄色油状物(化合物c)86.8g,收率62.1%;s3、合成下式的化合物d:[4
‑
(3
‑
氨基
‑
丙氨基)丁基]
‑
氨基甲酸叔丁酯,其分子式为:c
12
h
27
n3o2,分子量为:245.36,化学结构式如下:;其制备方法包括如下步骤:将化合物c(78g,0.3mol)溶于甲苯300ml和四氢呋喃300ml的混合溶剂中,氮气保护,冰浴下控温0
±
5℃滴加70%红铝甲苯溶液(173.3g,0.6mol),加完后继续保温搅拌2小时,升温至80℃,保温20h,冰浴下滴入25%氯化铵水溶液500ml淬灭反应,过滤后分液,水相减压脱干后加入200ml甲醇,充分打散后过滤,滤液减压蒸干得到化合物d;s4、合成下式的化合物e:n
‑
(3
‑
氨基丙基)
‑
1,4
‑
丁二胺,即亚精胺,其分子式为:c7h
19
n3,分子量为:145.25,化学结构式如下:;其制备方法包括如下步骤:将上一步化合物d(0.3mol)溶于450ml甲醇中,冰水浴中滴入盐酸(250ml,3mol),搅拌4h,减压蒸干,用碳酸钾固体调至ph=11~12,40℃搅拌2小时后,减压脱干,加入四氢呋喃300ml萃取,过滤,滤液浓缩干,减压精馏,于12mmhg真空下收集124~126℃馏分,得到亚精胺29.8g,收率69.4%,气相纯度98.5%。
[0037]
实施例5:s1、合成下式的化合物b:1
‑
(叔丁氧基羰基)
‑2‑
吡咯烷酮,其分子式为:c9h
15
no3,
分子量为:185.22,化学结构式如下:;其制备方法包括如下步骤:将吡咯烷酮(255.1g,3.0mol,1.0eq),乙腈2.0l,4
‑
二甲氨基吡啶(45.81g,0.375mol,0.125eq)加入3l反应瓶中,水浴中滴加二碳酸二叔丁酯(713.6g,3.27mol,1.09eq),10~20℃反应2h,40℃减压浓缩干,加入1175g甲基叔丁基醚溶解,用137g柠檬酸/1400ml水溶液,40g碳酸氢钠/600ml水溶液,40g氯化钠/600ml水溶液洗后,有机相脱除溶剂,经后处理提纯后得到浅黄色油状物化合物b共534.5g,收率96.2%;s2、合成下式的化合物c:[3
‑
(3
‑
氨基
‑
丙氨甲酰基)丙基]
‑
氨基甲酸叔丁酯,其分子式为:c
12
h
25
n3o3,分子量为:259.35,化学结构式如下:;其制备方法包括如下步骤:将1,3
‑
丙二胺(60.0g,0.81mol,1.5eq),化合物b(100g,0.54mol,1.0eq)以及四氢呋喃500ml混合,于室温搅拌30h,减压脱除溶剂和过量的1,3
‑
丙二胺,剩余体系加入400ml水中,用柠檬酸调至ph=5,醋酸异丙酯 100ml萃取3次,水层加入60g氯化钠,用30%氢氧化钠水溶液调至ph=7,水相用二氯甲烷150ml*4萃取,脱除溶剂,得到黄色油状物(化合物c)89.9g,收率64.3%;s3、合成下式的化合物d:[4
‑
(3
‑
氨基
‑
丙氨基)丁基]
‑
氨基甲酸叔丁酯,其分子式为:c
12
h
27
n3o2,分子量为:245.36,化学结构式如下:;其制备方法包括如下步骤:将化合物c(78g,0.3mol)溶于四氢呋喃1000ml,氮气保护,冰浴下0
±
5℃滴加50%硼烷四氢呋喃溶液(168g,0.6mol),升至30℃,保温16h,冰浴下滴入甲醇600ml淬灭反应,45℃水浴减压浓缩干,得化合物d;s4、合成下式的化合物e:n
‑
(3
‑
氨基丙基)
‑
1,4
‑
丁二胺,即亚精胺,其分子式为:c7h
19
n3,分子量为:145.25,化学结构式如下:;其制备方法包括如下步骤:将上一步化合物d(0.3mol)溶于450 ml甲醇中,冰水浴中滴入盐酸(250ml,3mol),搅拌4h,减压蒸干,用碳酸钾固体调至ph=11~12,40℃搅拌2小时后,减压脱干,加入四氢呋喃300ml萃取,过滤,滤液浓缩干,减压精馏,于12mmhg真空下收集124~126℃馏分,得到亚精胺31.1g,收率72.4%,气相纯度98.9%。
[0038]
实验例:以某新材料公司利用上述方法制备亚精胺的实际应用为例,该公司利用上述实施例分别制备亚精胺后将制备过程中的产品相关数据记录如下表中所示(注:步骤3未经提纯直接参与下一步步骤4的反应,因此步骤3和步骤4的摩尔收率为两步合并摩尔收率)。
专利申请号为cn201210138708.4专利共5个合成步骤,总收率30%,产品纯度为99.5%且涉及多步危险反应;专利申请号为cn201810833825.x专利共5个步骤总收率为35%,产品纯度为98%,多步涉及危险反应,且原料氨基丙醇价格较高。
[0039]
综上可以看出,本发明所使用原料吡咯烷酮、1,3
‑
丙二胺等均为常见化工原料,价格便宜易得,反应条件相对温和,整体反应中安全隐患较低;利用本方法制得产物的合并收率达到62.1%~79.5%,且成品亚精胺的纯度可达到99.6%,可以很好地解决现有技术中存在的问题。
[0040]
以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围,其均应涵盖在本发明的权利要求和说明书的范围当中;对于本技术领域的技术人员来说,对本发明实施方式所做出的任何替代改进或变换均落在本发明的保护范围内。本发明未详述之处,均为本技术领域技术人员的公知。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1