一种德拉沙星及其中间体的精制方法与流程
1.本发明属于医药合成领域,具体地,本发明提供了一种德拉沙星及其中间体的精制方法,其中包括使用一种硫酸的混合酸作为反应试剂。
背景技术:
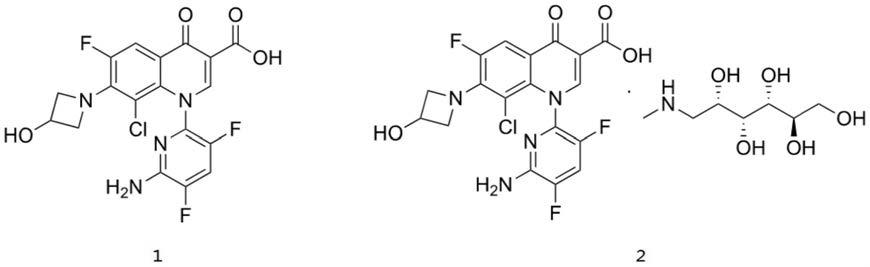
2.德拉沙星化学名为:1-(6-氨基-3,5-二氟吡啶-2-基)-8-氯-6-氟-7-(3-羟基氮杂环丁烷-1-基)-4-氧代-1,4-二氢喹啉-3-羧酸,结构式如1所示,其相应的葡甲胺盐结构如式2所示:
[0003][0004]
德拉沙星是最新一代广谱氟喹诺酮抗菌药物,最开始由日本wakunaga制药公司研发,后授权给美国melinta制药公司进行开发,并在2017年6月19日由美国fda批准上市。其主要用于治疗由易感细菌引起的急性细菌性皮肤和皮肤结构感染。德拉沙星与其他氟喹诺酮类抗菌药物相比,具有更优异的抗革兰氏阳性菌活性,特别是对其他喹诺酮类抗菌药耐药的甲氧西林耐药金黄色葡萄球菌。另外,德拉沙星能在酸性条件下保持其活性,这一特性能更有效地消灭酸性环境中的金黄色葡萄球菌,因此在治疗皮肤感染、呼吸道感染、泌尿系统感染等方面发挥重要作用。
[0005]
对于德拉沙星的合成方法,目前主要存在两种方式:
[0006]
第一种方法(wo1997011068a1):
[0007][0008]
本方法是由日本wakunaga制药公司以3-氯-2,4,5-三氟苯甲酸为起始物料经过多步反应得到德拉沙星,但是该起始物料价格高昂不易购买,且反应合成步骤中经过高温环合和高温水解,产生杂质过多,造成产品纯度降低,不适合于工业化生产。
[0009]
第二种方法(wo2006015194a2):
[0010][0011]
本方法是由美国abbott制药公司以2,4,5-三氟苯甲酸为起始物料进行反应合成,但其中关键的步骤即倒数第二步氯化反应,会造成杂质增多,且后一步水解反应温度达到50℃,温度高同样也会造成产物杂质增多,造成产物纯度降低。更不幸的是,反应完成分离出德拉沙星产物后,经hplc检测,在相对保留时间为1.60时出现一个面积高达0.43%的杂质,该杂质即使在最后成盐反应中也是难以清除的,这就使得该方法合成的德拉沙星葡甲胺难以达到药用级别。hanselmann,r.等人的《《identification and suppression of a dimer impurity in the development of delafloxacin》》,organic process research&development,vol.13,pages 54-59(2009)中对该杂质进行鉴别,经过实验验证它被鉴定为德拉沙星的二聚物,其结构式如3所示:
[0012]
[0013]
并在该文中指出该杂质产生的主要原因,是由于氯化反应中使用了硫酸试剂,造成了式a-7化合物中的氮杂环丁烷环开环,进而在后一步水解反应完成之后产生了该杂质。
[0014]
cn102164912b公开了一种制备德拉沙星化合物的工艺,并且使其中产生的德拉沙星二聚物杂质的含量低于0.40%。虽然说这个方法可以使德拉沙星二聚物杂质的量得到有效控制,但是仍然不适合于放大工业化生产,因此,需要寻求更优化的方法以彻底减少该二聚物杂质的生产,进一步提高产物的纯度。
技术实现要素:
[0015]
本发明的目的是,提供一种制备德拉沙星及其中间体的方法,该方法在进行大规模生产时可以彻底抑制德拉沙星二聚物杂质的产生,获得超高纯度,高收率的德拉沙星,满足药用要求,适合放大工业化生产。
[0016]
根据上述德拉沙星二聚物杂质产生的原因,本发明人尝试对由式a-6化合物生成德拉沙星的氯化反应步骤和水解反应步骤进行研究。本发明人惊奇的发现,当使用硫酸的混合酸作为氯化反应的反应试剂时,可以完全避免德拉沙星二聚物杂质的产生且最终获得了超高纯度,高收率的德拉沙星。
[0017]
为实现本发明的目的,提供了式a-7化合物的制备方法:
[0018]
将式a-6化合物与氯化试剂和硫酸的混合酸进行反应得到式a-7化合物,
[0019]
反应式如下式所示:
[0020][0021]
优选的,所述氯化试剂为n-氯代琥珀酰亚胺。
[0022]
优选的,所述硫酸的混合酸为硫酸与有机酸的混合酸,所述有机酸选自柠檬酸、苦味酸、苹果酸、草酸和酒石酸中的一种或几种,优选为硫酸与柠檬酸的混合酸。示例性的,如硫酸和柠檬酸的混合酸为在室温下,将硫酸和柠檬酸在乙酸甲酯溶液中进行混合制得。
[0023]
优选的,所述氯化试剂与式a-6化合物的摩尔比为(1-2):1,进一步优选的,式a-6化合物的摩尔比为(1.16-1.20):1,更为优选的,式a-6化合物的摩尔比为1.18:1。
[0024]
优先的,所述硫酸的混合酸与中间体i的摩尔比为(0.005-0.1):1,进一步优选的为(0.01-0.05):1,更为优选为(0.016-0.02):1;其中硫酸与所述有机酸的摩尔比为≥2,更为优选的硫酸与所述有机酸的摩尔比为3。
[0025]
优选的,反应溶剂选自二甲基亚砜、乙醇、甲醇、四氢呋喃、二氯乙烷、乙酸乙酯、乙酸甲酯、乙腈、丙酮、正庚烷、正己烷以及其中两种或两种以上的组合物,进一步优选为乙酸乙酯。
[0026]
优选的,所述的反应温度控制在10-35℃。
[0027]
进一步的,所述式a-7化合物经过水解反应得到式1化合物,反应式如下式所示:
[0028][0029]
上述水解反应选用氢氧化物碱为水解试剂,其中氢氧化物碱选自氢氧化锂、氢氧化钠、氢氧化钾、氢氧化铯以及其中两种或两种以上的组合物,优选氢氧化钾。
[0030]
上述所述水解反应的温度控制10-35℃。
[0031]
进一步的,本发明提供一种德拉沙星的检测方法:
[0032]
照高效液相色谱法,以十八烷基硅烷键合硅胶为填充剂的色谱柱;流动相a为80%甲醇乙腈溶液,流动相b为50mm的乙酸铵溶液,检测波长为290nm,流速为每分钟0.5ml,柱温30℃,按照下表1进行梯度洗脱:
[0033]
表1
[0034]
时间(分钟)流动相a(%)流动相b(%)03565173862204060305050496535559010603565
[0035]
按照以上提供的hplc检测方法,以最终合成的德拉沙星作为供试品,可以检测到德拉沙星的含量以及德拉沙星二聚物等杂质的含量。
[0036]
本发明的有益效果
[0037]
本发明提供了一种德拉沙星及其中间体的精制方法,与现有公开的德拉沙星的精制方法相比,本发明的技术方案可以彻底的避免德拉沙星二聚物杂质的生成。除此之外,本发明由式a-7化合物生成德拉沙星的水解反应温度为10-35℃,与现有公开的德拉沙星合成方法中的水解反应温度都高达50℃以上相比,在得到相同效果的情况下,该反应温度更加的温和,更利于工艺操作且进一步减少杂质的产生,使反应得到超高纯度、高收率的德拉沙星,德拉沙星的纯度可以到达99.6%以上,更适用于放大工业化生产。
附图说明
[0038]
图1示出了德拉沙星二聚物杂质的hplc定位图谱。
[0039]
图2示出了制备工业规模量的德拉沙星的hplc检测图谱。
具体实施方式
[0040]
为了更好地理解本发明的技术方案,下面结合具体的实施例对本发明的技术方案做进一步说明,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
[0041]
实施例1,德拉沙星的制备
[0042]
向1l反应瓶中加入50.0g式a-6化合物和150ml乙酸乙酯,控温10-35℃。滴加ncs溶液(通过将15.6gncs加入到0.13g硫酸与0.09g柠檬酸的250ml乙酸甲酯的混合溶液中完成),滴加完毕后反应6-10小时后,分别用250g1.5%碳酸氢钠水溶液、140g10%亚硫酸钠水溶液洗涤分液,真空蒸发溶剂得到51.0g式a-7化合物,收率95.7%。之后向反应瓶中加入30g式a-7化合物、240g异丙醇以及氢氧化钾水溶液(9.1g氢氧化钾溶于225g水),控温10-35℃反应3h后,加入12%乙酸溶液143g,搅拌1h后抽滤干燥得23.7g德拉沙星,收率96.1%,纯度99.9%,未检测到德拉沙星二聚物杂质。
[0043]
实施例2,德拉沙星的制备
[0044]
向1l反应瓶中加入50.0g式a-6化合物和150ml乙酸乙酯,控温10-35℃。滴加ncs溶液(通过将15.6gncs加入到0.04g硫酸与0.26g柠檬酸的250ml乙酸甲酯的混合溶液中完成),滴加完毕后反应6-10小时后,分别用250g1.5%碳酸氢钠水溶液、140g10%亚硫酸钠水溶液洗涤分液,真空蒸发溶剂得到49.6g式a-7化合物,收率93%。之后向反应瓶中加入30g式a-7化合物、240g异丙醇以及氢氧化钾水溶液(9.1g氢氧化钾溶于225g水),控温10-35℃反应3h后,加入12%乙酸溶液143g,搅拌1h后抽滤干燥得22.1g德拉沙星,收率89.8%,纯度99.0%,未检测到德拉沙星二聚物杂质。
[0045]
实施例3,德拉沙星的制备
[0046]
向1l反应瓶中加入50.0g式a-6化合物和150ml乙酸乙酯,控温10-35℃。滴加ncs溶液(通过将15.6gncs加入到0.34g柠檬酸的250ml乙酸甲酯的混合溶液中完成),滴加完毕后反应6-10小时后,分别用250g1.5%碳酸氢钠水溶液、140g10%亚硫酸钠水溶液洗涤分液,真空蒸发溶剂得到48.5g式a-7化合物,收率91.0%。之后向反应瓶中加入30g式a-7化合物、240g异丙醇以及氢氧化钾水溶液(9.1g氢氧化钾溶于225g水),控温10-35℃反应3h后,加入12%乙酸溶液143g,搅拌1h后抽滤干燥得20.0g德拉沙星,收率81.1%,纯度92.1%。
[0047]
实施例4,德拉沙星的制备
[0048]
向1l反应瓶中加入50.0g式a-6化合物和150ml乙酸乙酯,控温10-35℃。滴加ncs溶液(通过将15.6gncs加入到0.13g硫酸与0.07g酒石酸的250ml乙酸甲酯的混合溶液中完成),滴加完毕后反应6-10小时后,分别用250g1.5%碳酸氢钠水溶液、140g10%亚硫酸钠水溶液洗涤分液,真空蒸发溶剂得到49.6g式a-7化合物,收率93%。之后向反应瓶中加入30g式a-7化合物、240g异丙醇以及氢氧化钾水溶液(9.1g氢氧化钾溶于225g水),控温10-35℃反应3h后,加入12%乙酸溶液143g,搅拌1h后抽滤干燥得22.5g德拉沙星,收率91.6%,纯度98.8%,未检测到德拉沙星二聚物杂质。
[0049]
实施例5,德拉沙星的制备,工业规模级别的投量,即kg级别以上。
[0050]
向1000l反应瓶中加入50kg式a-6化合物和150l乙酸乙酯,控温10-35℃。滴加ncs溶液(通过将15.6kgncs加入到130g硫酸与90g柠檬酸的250l乙酸甲酯的混合溶液中完成),滴加完毕后反应6-10小时后,分别用250kg1.5%碳酸氢钠水溶液、140kg10%亚硫酸钠水溶液洗涤分液,真空蒸发溶剂得到50.5kg式a-7化合物,收率94.6%。之后向反应瓶中加入30kg式a-7化合物、240kg异丙醇以及氢氧化钾水溶液(9.1kg氢氧化钾溶于225kg水),控温10-35℃反应3h后,加入12%乙酸溶液143kg,搅拌1h后抽滤干燥得23.9kg德拉沙星,收率96.9%,纯度99.6%,未检测到德拉沙星二聚物杂质。德拉沙星二聚物杂质的hplc的定位谱
图如图1所示,其对应的谱图数据如下表2;制备的德拉沙星hplc检测图谱如图2所示,其对应的谱图数据如下表3。
[0051]
表2
[0052]
[0053]
表3
[0054][0055]
对比例1,德拉沙星的制备向1l反应瓶中加入50.0g式a-6化合物和150ml乙酸乙酯,控温10-35℃。滴加ncs溶液(通过将15.6gncs加入到0.17g硫酸的250ml乙酸甲酯的混合溶液中完成),滴加完毕后反应6-10小时后,分别用250g1.5%碳酸氢钠水溶液、140g10%亚硫酸钠水溶液洗涤分液,真空蒸发溶剂得到46.1g式a-7化合物,收率86.3%。之后向反应瓶中加入30g式a-7化合物、240g异丙醇以及氢氧化钾水溶液(9.1g氢氧化钾溶于225g水),控温10-35℃反应3h后,加入12%乙酸溶液143g,搅拌1h后抽滤干燥得22.1g德拉沙星,收率89.7%,纯度94.2%,检测到0.3%的德拉沙星二聚物杂质。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1