一种替米沙坦关键中间体4的制作方法
一种替米沙坦关键中间体4
′-甲基联苯-2-羧酸酯的制备方法
技术领域
1.本发明属于药物制备技术领域,涉及一种替米沙坦关键中间体4
’‑
甲基联苯-2-羧酸酯的制备方法。
背景技术:
2.替米沙坦是一种长效、高效、低毒的新型at拮抗剂,系由德国贝林格尔-因格海姆药厂开发,并于1997年上市。它也是一个非肽类血管紧张素ⅱ受体拮抗剂,可选择性地、难以逆转的阻滞at1受体,而对其他受体系统尤其是涉及心血管系统的受体无影响。
3.替米沙坦化学名称为:4
′‑
[4-甲基-6-(1-甲基-1h-苯并咪唑-2-基)-2-丙基-1h-苯并咪唑-1
‑ꢀ
基甲基]联苯基-2-羧酸,其化学结构式如下:
[0004][0005]4’‑
甲基联苯-2-羧酸酯为替米沙坦的关键中间体。目前,4
’‑
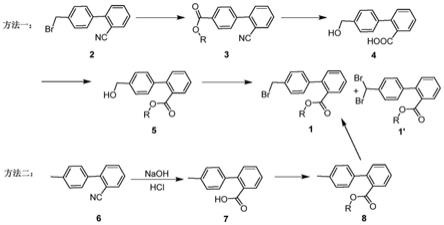
甲基联苯-2-羧酸酯的制备方法主要有以下几种(图1):
[0006]
方法一:中国专利cn101172953a中采用4
′‑
溴甲基-2-氰基联苯为起始原料,经酯化、水解、再酯化、最后溴化反应制得。
[0007]
方法二:中国专利申请cn113105327a和中国专利申请cn105399627a均采用2-氰基
‑ꢀ4’‑
甲基联苯为起始原料,经水解、酯化、溴代制备得4
’‑
溴甲基-2-联苯羧酸甲酯。
[0008]
方法一路线较长,且起始物料采用溴甲基氰基联苯,先脱溴后面又上溴,增加了生产步骤且三废较多。方法二采用先水解后酯化再酯化的方法,虽然相对方法一路线较短,但水解和酯化分开进行,实际操作也较为繁琐。此外,不论是方法一还是方法二,最后的溴代反应以低沸点的二氯甲烷为反应溶剂,采用nbs或溴化氢在光照条件催化进行自由基反应,由于自由基反应难以控制且反应体系为二相,反应中二溴副产物(化合物1
′
)较多,而目标产物 (化合物1)和副产物(化合物1
′
)极性极为相似,难以精制清除,导致目标产物纯度≤98.5%,精制损失≥10%,溴代步骤收率≤85%,总摩尔收率低≤70%,成本相对较高,三废较多。
[0009]
因此,本领域亟需一种收率高、副产物少、成本低、更适用于工业化生产的替米沙坦中间体的制备方法。
技术实现要素:
[0010]
本发明的目的在于解决现有制备方法中4
’‑
甲基联苯-2-羧酸酯收率低、原料转化率低或副产物高,操作复杂,成本高,不适用于工业化生产等缺陷,提供一种替米沙坦中间
联苯羧酸正丁酯、 4
′‑
溴甲基-2-联苯羧酸异丁酯。
[0031]
具体的,一种替米沙坦关键中间体4
′‑
溴甲基-2-联苯羧酸酯的制备方法,包括如下步骤:
[0032]
步骤(1)、将4
′‑
甲基-2-氰基联苯和醇类化合物混合,升温至80℃,加入酸,常压或加压下,在温度100~150℃下进行水解、酯化反应,直至2-氰基-4
′‑
甲基联苯反应≤0.1%;减压浓缩至醇类化合物为醇类化合物初始用量的8%~35%wt,避免全部蒸出溶剂导致釜底结块,同时,保留部分溶剂可以保证生产中容易搅拌,降温至≤15℃,加入与醇类化合物初始用量等量的水,室温搅拌1-2h,过滤,干燥至恒重,得到4
′‑‑
甲基-2-联苯羧酸酯纯品;
[0033]
步骤(2)、以有机溶剂为反应溶剂,4
′‑
甲基-2-联苯羧酸酯与双氧水、溴源在相转移催化剂下,在回流条件下进行溴代反应;反应结束后降温至≤30℃,加入10%亚硫酸氢钠溶液淬灭反应,搅拌,过滤,滤饼用与反应溶剂相同的有机溶剂润洗,干燥,得到4
′‑
溴甲基-2
‑ꢀ
联苯羧酸酯。
[0034]
本发明的有益效果:
[0035]
(1)、与现有工艺中水解、酯化分两步或多步操作相比,本发明“一锅法”水解、酯化反应具有操作简单、三废少、绿色环保的优势。
[0036]
(2)、本发明溴代反应使用极性较小沸点相对较高的溶剂,通过控制反应温度使得自由基反应更易进行;通过添加催化剂,更多的原料参与溴代反应,避免目标物4
’‑
甲基联苯-2
‑ꢀ
羧酸酯进一步溴代产生二溴副产物(化合物1
′
),从而有效提高原料的转化率同时减少二溴副产物的产生;本发明方法操作简单、专一性高、副产物少,能将4
’‑
甲基联苯-2-羧酸酯残留控制在0.1%以下,4
′
,4
′‑
二溴甲基联苯-2-羧酸酯控制在0.1%以下,最终在无需精制的条件下可制得纯度纯度99.3%以上的目标产物,总摩尔收率达90%以上,适合工业化生产。
附图说明
[0037]
图1为现有4
’‑
甲基联苯-2-羧酸酯的反应路线图。
具体实施方式
[0038]
下面通过实施例式进一步说明本发明的技术方案,但并不应因此将本发明限制在所述的实施例范围内。下列实施例中未注明具体实施条件的,按照常规方法和条件。
[0039]
实施例1
[0040]
往1l反应瓶中加入2-氰基-4
′‑
甲基联苯38.7g(0.2mol)和116g正丁醇,升温至80℃,边搅拌边滴加40.0g(0.4mol)浓硫酸(质量分数98%,下同);滴毕,升温至118℃回流反应,hplc中控,4h后,2-氰基-4
′‑
甲基联苯反应≤0.1%,停止反应;减压浓缩至剩余正丁醇为38g左右,降温至≤15℃,往体系中加入116g纯化水;加毕,室温搅拌1.5h,过滤,滤饼 50℃减压烘干至恒重,得到4
′‑
甲基-2-联苯羧酸正丁酯(白色固体)50.7g,摩尔收率94.5%。
[0041]4′‑
甲基-2-联苯羧酸正丁酯40.3g(0.15mol)加入到200g正己烷和33.4g质量分数40%氢溴酸(0.165mol)中,再加入0.05g(0.15mmol)tbab;加毕,在搅拌下升温至60℃,开始滴加质量分数30%的双氧水17.0g(0.15mol),在2h左右内滴毕,升温至回流反应 30min,hplc中控至原料残留≤1.0%;降温至≤30℃,滴加10%亚硫酸氢钠溶液53g,滴毕,搅拌
30min,过滤,滤饼用30g正己烷润洗,滤饼烘干,得到4
′‑
溴甲基-2-联苯羧酸正丁酯(白色固体)49.8g,摩尔收率95.6%,hplc:99.43%,二溴杂质未检出。
[0042]
实施例2
[0043]
往1l反应瓶中加入2-氰基-4
′‑
甲基联苯38.7g(0.2mol)和116g异丁醇,升温至80℃,边搅拌边滴加78.4g(0.784mol)浓硫酸;滴毕,升温至105℃回流反应,hplc中控,6h 后,2-氰基-4
′‑
甲基联苯反应≤0.1%,停止反应;减压浓缩至剩余异丁醇为38g左右,降温至≤15℃,往体系中加入116g纯化水;加毕,室温搅拌1.5h,过滤,滤饼50℃减压烘干至恒重,得到4
′‑
甲基-2-联苯羧酸异丁酯(白色固体)50.5g,摩尔收率94.2%。
[0044]4′‑
甲基-2-联苯羧酸异丁酯40.3g(0.15mol)加入到200g正己烷和33.4g40%氢溴酸 (0.165mol)中,再加入0.15g(0.45mmol)tbab;加毕,在搅拌下升温至60℃,开始滴加30%的双氧水34.0g(0.30mol),在2h左右内滴毕,升温至回流反应30min,hplc中控至原料残留≤1.0%;降温至≤30℃,滴加10%亚硫酸氢钠溶液53g,滴毕,搅拌30min,过滤,滤饼用30g正己烷润洗,滤饼烘干,得到4
′‑
溴甲基-2-联苯羧酸异丁酯(白色固体)50.6g,摩尔收率97.2%,hplc:99.58%,二溴杂质未检出。
[0045]
实施例3
[0046]
往1l反应瓶中加入2-氰基-4
′‑
甲基联苯38.7g(0.2mol)和194g正丁醇,升温至80℃,边搅拌边滴加40.0g(0.4mol)浓硫酸;滴毕,体系在5-10mpa下控制温度至150℃反应, hplc中控,1h后,2-氰基-4
′‑
甲基联苯反应≤0.1%;停止反应,减压浓缩至剩余正丁醇为 37g左右,降温至≤15℃,往体系中加入116g纯化水;加毕,室温搅拌1.5h,过滤,滤饼 50℃减压烘干至恒重,得到4
′‑
甲基-2-联苯羧酸正丁酯(白色固体)51.1g,摩尔收率95.3%。
[0047]4′‑
甲基-2-联苯羧酸正丁酯40.3g(0.15mol)加入到200g正己烷和33.4g40%氢溴酸(0.165mol)中,再加入0.125g(0.45mmol)tbac;加毕,在搅拌下升温至60℃,开始滴加30%的双氧水34.0g(0.30mol),在2h左右内滴毕,升温至回流反应30min,hplc中控至原料残留≤1.0%;降温至≤30℃,滴加10%亚硫酸氢钠溶液53g,滴毕,搅拌30min,过滤,滤饼用30g正己烷润洗,滤饼烘干,得到4
′‑
溴甲基-2-联苯羧酸正丁酯(白色固体)50.5g,摩尔收率97.0%,hplc:99.53%,二溴杂质未检出。
[0048]
实施例4
[0049]
往1l加压反应釜中加入2-氰基-4
′‑
甲基联苯58.0g(0.3mol)和580g乙醇,升温至 75℃,边搅拌边滴加45.0g(0.45mol)浓硫酸,滴毕,体系在5-10mpa下控制温度在100℃反应,16h后开始hplc中控,此后每隔1h中控一次,至2-氰基-4
′‑
甲基联苯反应≤0.1%;体系减压浓缩至剩余乙醇为50g左右,降温至≤15℃,往体系中加入174g纯化水;加毕,室温搅拌1.5h,过滤,滤饼45℃减压烘干至恒重,得到4
′‑
甲基-2-联苯羧酸乙酯(白色固体)68.6g,摩尔收率95.1%。
[0050]4′‑
甲基-2-联苯羧酸乙酯48g(0.2mol)加入到240g乙酸乙酯和41.8g40%氢溴酸 (0.2mol)中,再加入0.137g(0.6mmol)teba;加毕,在搅拌下升温至60℃,开始滴加 30%的双氧水68.0g(0.6mol),4h左右滴毕,升温至回流反应30min,hplc中控至原料残留≤1.0%;降温至≤30℃,滴加10%亚硫酸氢钠溶液416g,滴毕,搅拌30min,过滤,滤饼用≤15℃的乙酸乙酯30g润洗,滤饼烘干,得到4
′‑
溴甲基-2-联苯羧酸乙酯(白色固体)62.0g,摩尔收率97.1%,hplc:99.69%,二溴杂质未检出。
[0051]
实施例5
[0052]
往1l加压反应瓶中加入2-氰基-4
′‑
甲基联苯58g(0.3mol)和406g甲醇,升温至 60℃,边搅拌边滴加45.0g(0.45mol)浓硫酸;滴毕,体系在5-10mpa下控制温度在110℃反应,12h后开始hplc中控,此后每隔1h中控一次,至2-氰基-4
′‑
甲基联苯反应≤0.1%;体系减压浓缩至剩余甲醇为50g左右,降温至≤15℃,往体系中加入174g纯化水;加毕,室温搅拌1.5h,过滤,滤饼45℃减压烘干至恒重,得到4
′‑
甲基-2-联苯羧酸甲酯(白色固体)64.6g,摩尔收率95.2%。
[0053]4′‑
甲基-2-联苯羧酸甲酯45.3g(0.2mol)加入到275g石油醚中,再加入0.32g (1mmol)tbab,在搅拌下升温至60℃,同时滴加30%的双氧水22.7g(0.2mol)和溴素 16.0g(0.1mol),在2h左右内滴毕,升温至回流反应30min,hplc中控至原料残留≤1.0%;降温至≤30℃,滴加10%亚硫酸氢钠溶液70g,滴毕,搅拌30min,过滤,滤饼用 150g石油醚润洗,滤饼烘干,得到4
′‑
溴甲基-2-联苯羧酸甲酯(白色固体)58.2g,摩尔收率 95.3%,hplc:99.40%,二溴杂质未检出。
[0054]
实施例6
[0055]
往1l加压反应瓶中加入2-氰基-4
′‑
甲基联苯58g(0.3mol)和174g甲醇,在温度5
‑ꢀ
15℃边搅拌边通入16.4g(0.45mol)氯化氢,体系在5-10mpa下升温至110℃反应,12h开始hplc中控,此后每隔1h中控一次,至2-氰基-4
′‑
甲基联苯反应≤0.1%;体系减压浓缩至剩余甲醇为50g左右,降温至≤15℃,往体系中加入174g纯化水;加毕,室温搅拌1.5h,过滤,滤饼45℃减压烘干至恒重,得到4
′‑
甲基-2-联苯羧酸甲酯(白色固体)64.4g,摩尔收率 94.9%。
[0056]4′‑
甲基-2-联苯羧酸甲酯45.3g(0.2mol)加入到275g正己烷中,再加入36.3g (0.204mol)nbs和0.32g(1mmol)tbab,在搅拌下升温至60℃,开始滴加30%的双氧水22.7g(0.2mol),在2h左右内滴毕,升温至回流反应30min,hplc中控至原料残留≤1.0%;降温至≤30℃,滴加10%亚硫酸氢钠溶液70g,滴毕,搅拌30min,过滤,滤饼用 150g正己烷润洗,滤饼烘干,得到4
′‑
溴甲基-2-联苯羧酸甲酯(白色固体)58.0g,摩尔收率 94.9%,hplc:99.43%,二溴杂质未检出。
[0057]
实施例7
[0058]
往1l加压反应瓶中加入2-氰基-4
′‑
甲基联苯116g(0.6mol)和580g甲醇,升温至 60℃,边搅拌边滴加243.4g(2.4mol)浓盐酸(质量分数36%);滴毕,体系在5-10mpa下控制温度在110℃反应,12h开始hplc中控,此后每隔1h中控一次,至2-氰基-4
′‑
甲基联苯反应≤0.1%;体系减压浓缩至剩余甲醇为80g左右,降温至≤15℃,保温搅拌1.5h,过滤,滤饼45℃减压烘干至恒重,得到4
′‑
甲基-2-联苯羧酸甲酯(白色固体)127.7g,摩尔收率 94.1%。
[0059]4′‑
甲基-2-联苯羧酸甲酯45.3g(0.2mol)加入到275g正己烷中,再加入36.3g (0.204mol)nbs和1.64g(0.01mol)aibn,在搅拌下升温至60℃,开始滴加30%的双氧水45.4g(0.4mol),在2h左右内滴毕,升温至回流反应30min,hplc中控至原料残留≤1.0%;降温至≤30℃,滴加10%亚硫酸氢钠溶液70g,滴毕,搅拌30min,过滤,滤饼用 150g正己烷润洗,滤饼烘干,得到4
′‑
溴甲基-2-联苯羧酸乙酯(白色固体)58.8g,摩尔收率 96.2%,hplc:99.54%,二溴杂质未检出。
[0060]
对比例1
[0061]
将4
′‑
甲基-2-联苯羧酸甲酯45.3g(0.2mol,实施例7)加入到275g正己烷中,再加入 36.3g(0.204mol)nbs,升温至60℃,开始滴加30%的双氧水45.4g(0.4mol),在2h左右内滴毕,升温至回流反应30min,hplc中控原料残留14.09%(反应时间同实施例7);降温至≤30℃,滴加10%亚硫酸氢钠溶液70g,滴毕,搅拌30min,过滤,滤饼用150g正己烷润洗,滤饼烘干,得到4
′‑
溴甲基-2-联苯羧酸乙酯(白色固体)51.5g,摩尔收率84.2%, hplc:96.05%,原料残留为1.42%,二溴杂质为1.34%。
[0062]
对比例2
[0063]
采用专利cn105399627a实施例1的方法制得的4
′‑
甲基-2-甲酸酯联苯(即4
′‑
甲基-2-联苯羧酸甲酯,下同)为原料,进行溴代反应:
[0064]
向反应瓶中加入27.7g(0.12mol)4
′‑
甲基-2-甲酸酯联苯、138.5g二氯甲烷、24g (0.13mol)nbs,在18000lx光照条件下,25℃反应5h,hplc中控,此时原料残留 9.4%,二溴生成5.0%;加入10%亚硫酸氢钠溶液100g水洗有机层,再分别用饱和食盐水 100g、纯化水100g洗涤有机层,最后加入无水硫酸钠干燥有机层;干燥完后过滤,减压浓缩至干;加入22g甲醇,快速降温至0℃,搅拌析晶5h,只有少量固体析出;过滤,滤饼 35℃真空干燥8h,得到31.2g白色结晶,摩尔收率83.6%,hplc:97.75%,二溴残留: 1.78%。
[0065]
对比例3
[0066]
采用专利cn113105327a实施例1的方法制得的4
′‑
甲基-2-甲酸酯联苯为原料,进行溴代反应:
[0067]
反应瓶中加入25g(0.094mol)4
′‑
甲基-2-甲酸酯联苯、120g二氯甲烷和64g水,搅拌均匀;在80w的led灯源辐照下,并保持体系温度在25℃,同步滴加12g溴素(0.075mol) 和22.4g双氧水(质量分数30%),滴加完后,在25℃下保温反应5min,hplc中控,原料残留1.9%,二溴生成14.2%;停止反应,静置30min分层,有机层用饱和无水硫酸钠水溶液100g洗2次;有机层40℃减压浓缩(避免70℃减压过于剧烈存在安全隐患)至干;加入到50ml乙醇中,升温至50℃并保温30min、再降温0~5℃并保温1h,过滤,滤饼烘干得到27.5g白色结晶,摩尔收率81.6%,hplc:97.3%,二溴残留:1.9%。
[0068]
对比例4
[0069]
将4
′‑
甲基-2-联苯羧酸甲酯45.3g(0.2mol,实施例7)加入到275g二氯甲烷中,再加入 36.3g(0.204mol)nbs和1.64g(0.01mol)aibn,升温至40℃(已回流),开始滴加30%的双氧水45.4g(0.4mol),在2h左右内滴毕,继续回流反应30min,hplc中控原料残留 10.49%;降温至≤30℃,滴加10%亚硫酸氢钠溶液70g,滴毕,搅拌30min。静置分层,有机层用饱和食盐水200g水洗,分层后有机层再用无水硫酸钠干燥,过滤,滤液浓缩至干,加入30g甲醇,降温0~5℃并保温搅拌5h,过滤,滤饼用150g正己烷润洗,滤饼烘干,得到4
′‑
溴甲基-2-联苯羧酸乙酯(白色固体)49.3g,摩尔收率80.6%,hplc:96.83%,原料残留为1.15%,二溴杂质为1.30%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1